Injection Leads to
Islet Regeneration in Autoimmune Diabetes
Study points to unexpected treatment for type 1 diabetes
A possible cure for insulin-dependent diabetes is in sight
following a major medical breakthrough. Scientists in the
![]()
|
Nonobese diabetic (NOD) mice are a model for type 1 diabetes
in humans. Treatment of NOD mice with end-stage disease by
injection of donor splenocytes and complete Freund's
adjuvant eliminates autoimmunity and permanently
restores normoglycemia. The return of endogenous
insulin secretion is accompanied by the reappearance of
pancreatic ß cells. We now show that live donor male
or labeled splenocytes administered to diabetic NOD females contain
cells that rapidly differentiate into islet and ductal epithelial
cells within the pancreas. Treatment with irradiated splenocytes
is also followed by islet regeneration, but at a slower
rate. The islets generated in both instances are persistent,
functional, and apparent in all NOD hosts with permanent
disease reversal.
Immunobiology Laboratory, Massachusetts General Hospital and
Harvard Medical School, Building 149, 13th Street, Room 3602,
Charlestown, MA 02129, USA.
* To whom correspondence should
be addressed. E-mail: faustman@helix.mgh.harvard.edu
The NOD mouse exhibits spontaneous
autoimmunity that causes diabetes through destruction
of insulin-secreting pancreatic islets. A lymphoid
cell–specific proteasome defect in these mice
interrupts the presentation of self antigens by major histocompatibility
complex (MHC) class I molecules that is required for
negative selection of autoreactive naïve T cells (1,
2).
The proteasome defect also impairs activation of the
transcription factor nuclear factor–![]() B in pathogenic memory T
cells, increasing their susceptibility to apoptosis
induced by tumor necrosis factor–
B in pathogenic memory T
cells, increasing their susceptibility to apoptosis
induced by tumor necrosis factor–![]() (TNF-
(TNF-![]() ) (3–5).
Reselection of peripheral autoimmune naïve T cells is
possible by the introduction of matched MHC class
I–self peptide complexes, whereas self-directed
autoimmune memory T cells can be reselected by treatment
with TNF-
) (3–5).
Reselection of peripheral autoimmune naïve T cells is
possible by the introduction of matched MHC class
I–self peptide complexes, whereas self-directed
autoimmune memory T cells can be reselected by treatment
with TNF-![]() or by the induction
of the endogenous TNF-
or by the induction
of the endogenous TNF-![]() with
complete Freund's adjuvant (CFA) (5,
6).
Simultaneous treatment of severely diabetic NOD mice
with both TNF-
with
complete Freund's adjuvant (CFA) (5,
6).
Simultaneous treatment of severely diabetic NOD mice
with both TNF-![]() and
normal splenocytes partially or fully matched for MHC
class I antigens thus restores self-tolerance and
eliminates T cells directed against islets, resulting
in permanent reversal of established diabetes (7).
This "cure" is accompanied by the reappearance of
insulin-secreting islets in the pancreas which can
control blood glucose concentration in an apparently
normal manner.
and
normal splenocytes partially or fully matched for MHC
class I antigens thus restores self-tolerance and
eliminates T cells directed against islets, resulting
in permanent reversal of established diabetes (7).
This "cure" is accompanied by the reappearance of
insulin-secreting islets in the pancreas which can
control blood glucose concentration in an apparently
normal manner.
The new pancreatic islets in such treated NOD mice might arise
from several sources, either endogenous or donor-derived
sources. Donor nonlymphoid cells administered to mice
or humans can undergo rare transdifferentiation events
(8–25),
although these findings remain controversial (26,
27).
Alternatively, the regenerated islet cells in NOD mice
might be the products of fusion between donor and host
cells, in a mouse model of liver damage (28,
29).
Such fusion events generate cells with marked chromosomal abnormalities
(30,
31).
To investigate the origin of the new pancreatic islet cells
in NOD mice, we examined the relative abilities of live
versus irradiated donor splenocytes to restore
normoglycemia (32).
We injected CFA and either live or irradiated male CByB6F1
mouse splenocytes into severely diabetic NOD females,
which were used to ensure the absence of visible
islets and insulitis that could obscure dead or dying
islets (table S1). We controlled blood glucose
concentration with a temporary (40-day) implant of syngeneic
islets under the capsule of one kidney, which improved
treatment efficacy. Similar to our previous data (7),
six (67%) of the nine NOD mice that received live
splenocytes remained normoglycemic after removal of
the islet implant (Fig.
1A). In contrast, none of the eight animals that
received irradiated splenocytes remained normoglycemic;
they all rapidly developed severe hyperglycemia. (See
supporting online text and fig. S1.) In another experiment, the
islet transplant was maintained for 120 days before graft removal,
to allow a longer period for islet regeneration. Of the
12 NOD mice that received live splenocytes, 11 (92%) remained
normoglycemic for >26 weeks after disease onset or
beyond 52 weeks of age. Moreover, 11 (85%) of the 13
animals that received irradiated splenocytes also
remained normoglycemic for >27 weeks after disease
onset or beyond 48 weeks of age (Fig.
1A, table S2). Both live and irradiated
splenocytes could thus effect permanent disease
elimination, and with a longer period of imposed normoglycemia
greatly increasing the frequency of functional islet
recovery in both groups.
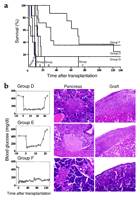
 Fig.
1. Effects of treatment with live or irradiated
splenocytes on the restoration of normoglycemia and
pancreatic histology in diabetic NOD mice. (A)
Kaplan-Meier plot for normoglycemia. Diabetic NOD females
were treated with a single injection of CFA and biweekly
injections for 40 days of either live (circles) or
irradiated (squares) splenocytes from CByB6F1
males. Syngeneic female islets transplanted subrenally at
the onset of treatment were removed after either 40 days
(left panel) or 120 days (right panel). Blood glucose
concentration was monitored at the indicated times after
islet graft removal, and the percentage of animals that
remained normoglycemic was plotted. Data are from 9 and 8
(left panel) or from 12 and 13 (right panel) animals that
received live or irradiated splenocytes, respectively; P
= 0.0002 (left panel), P = 0.68 (right panel) for
comparison between the two treatment groups. (B)
Pancreatic histology. Three NOD mice successfully treated
with either irradiated (top panels) or live (bottom
panels) splenocytes were killed Fig.
1. Effects of treatment with live or irradiated
splenocytes on the restoration of normoglycemia and
pancreatic histology in diabetic NOD mice. (A)
Kaplan-Meier plot for normoglycemia. Diabetic NOD females
were treated with a single injection of CFA and biweekly
injections for 40 days of either live (circles) or
irradiated (squares) splenocytes from CByB6F1
males. Syngeneic female islets transplanted subrenally at
the onset of treatment were removed after either 40 days
(left panel) or 120 days (right panel). Blood glucose
concentration was monitored at the indicated times after
islet graft removal, and the percentage of animals that
remained normoglycemic was plotted. Data are from 9 and 8
(left panel) or from 12 and 13 (right panel) animals that
received live or irradiated splenocytes, respectively; P
= 0.0002 (left panel), P = 0.68 (right panel) for
comparison between the two treatment groups. (B)
Pancreatic histology. Three NOD mice successfully treated
with either irradiated (top panels) or live (bottom
panels) splenocytes were killed |
Mice treated with irradiated splenocytes that exhibited
persistent normoglycemia for ![]() 9 weeks after nephrectomy (table S2)
exhibited the reappearance of pancreatic islets
without invasive insulitis (autoreactive cells within
the islets) but with pronounced peri-insulitis (circumferential
lymphoid cells that do not progress to invasion) (Fig.
1B, table S3). In contrast, the pancreas of NOD mice that
received live splenocytes exhibited the reappearance of
pancreatic islets without invasive insulitis and with
minimal or no peri-insulitis. The live splenocytes
were thus necessary for reduction of peri-insulitis but
not for the growth of new islets. Functionally, the restoration
of long-term normoglycemia was indistinguishable between
animals with disease reversal due to live or
irradiated splenocytes.
9 weeks after nephrectomy (table S2)
exhibited the reappearance of pancreatic islets
without invasive insulitis (autoreactive cells within
the islets) but with pronounced peri-insulitis (circumferential
lymphoid cells that do not progress to invasion) (Fig.
1B, table S3). In contrast, the pancreas of NOD mice that
received live splenocytes exhibited the reappearance of
pancreatic islets without invasive insulitis and with
minimal or no peri-insulitis. The live splenocytes
were thus necessary for reduction of peri-insulitis but
not for the growth of new islets. Functionally, the restoration
of long-term normoglycemia was indistinguishable between
animals with disease reversal due to live or
irradiated splenocytes.
We next tested mice that had been treated with live or
irradiated splenocytes for the presence of live donor
cells in blood, pancreas, and other tissues.
Peripheral blood lymphocytes (PBLs) from NOD mice
treated with irradiated CByB6F1 splenocytes showed
only background staining for H-2Kb (an indicator
of live donor cells), indicating that no donor
hematopoietic cells remained (Table
1; table S2 and fig. S2). In contrast, 4.4 to 12.6% of PBLs
from NOD mice treated with live CByB6F1 splenocytes
were of donor origin. PBLs from an untreated NOD mouse
contained only cells expressing H-2Kd, and
those from a CByB6F1 mouse contained
exclusively cells coexpressing H-2Kb and H-2Kd.
NOD mice treated with live splenocytes thus exhibited
a persistent low level of blood chimerism with
semiallogeneic cells that was achieved without
continuous immunosuppression or lethal preconditioning.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Flow cytometry also revealed between 3.5 and 4.7% of cells
positive for both H-2Kd and H-2Kb
among splenocytes from five NOD mice successfully
treated with live splenocytes; this confirmed the persistence
of donor CByB6F1 cells in all recipients (Table
1). Splenocytes from an untreated control NOD
mouse showed a background level of 0.3%
double-positive staining for both markers. CByB6F1
donor splenocytes also contributed to T cells (CD3+),
monocytes (CD11b), and B cells (CD45R+) (data
not shown).
We then examined parenchymal tissues for chimerism by
fluorescence in situ hybridization (FISH) analysis for
detection of the Y chromosome of the male donor cells
in two long-term normoglycemic NOD mice (Fig.
2A). Staining of serial pancreatic sections with antibodies
to insulin revealed a homogeneous insulin content in
the large islets (Fig.
2B; Table
1), consistent with the restored normoglycemia.
Single-color FISH analysis revealed abundant nuclei
positive for the Y chromosome within the islets (Fig.
2B; Table
1). In contrast, the exocrine portions of the pancreas
were largely devoid of male cells. In these five animals, 29
to 79% of islet cells were of donor origin. No islets solely
of host origin were detected.
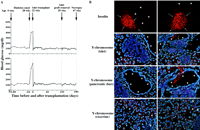 Fig.
2. Long-term restoration of normoglycemia and the
direct contribution of live donor splenocytes to islet
regeneration in successfully treated NOD female mice. (A)
Blood glucose concentrations during the lifetime of two
NOD females (top and bottom, nos. 789 and 790 in Table
1, respectively) successfully treated with CFA and
CByB6F1 male splenocytes, as well as with a
temporary subrenal transplant of syngeneic islets. (B)
Immunofluorescence and FISH analyses of serial pancreatic
sections from the successfully treated NOD females 789
(left) and 790 (right). The two top panels show
immunofluorescence staining of islets with antibodies to
insulin (red); the three pairs of images below show FISH
signals obtained with a Y chromosome–specific probe
(pink dots) and nuclear staining with DAPI (blue) in
sections containing islets (arrows), pancreatic ducts
(arrowheads), and exocrine pancreas, respectively. Fig.
2. Long-term restoration of normoglycemia and the
direct contribution of live donor splenocytes to islet
regeneration in successfully treated NOD female mice. (A)
Blood glucose concentrations during the lifetime of two
NOD females (top and bottom, nos. 789 and 790 in Table
1, respectively) successfully treated with CFA and
CByB6F1 male splenocytes, as well as with a
temporary subrenal transplant of syngeneic islets. (B)
Immunofluorescence and FISH analyses of serial pancreatic
sections from the successfully treated NOD females 789
(left) and 790 (right). The two top panels show
immunofluorescence staining of islets with antibodies to
insulin (red); the three pairs of images below show FISH
signals obtained with a Y chromosome–specific probe
(pink dots) and nuclear staining with DAPI (blue) in
sections containing islets (arrows), pancreatic ducts
(arrowheads), and exocrine pancreas, respectively. |
Male donor cells also contributed to the epithelium of NOD
female pancreatic ducts, although the distribution of
male cells in this tissue was more heterogeneous than
was that in islets (Fig.
2B, Table
1). Among the five treated NOD females studied in detail,
33 to 75% of the ducts contained genetic material of male
origin. Ducts purely of host origin were never associated with
an adjacent islet containing male cells. The proportion of
male cells in the pancreatic ducts of the five NOD mice ranged
from 9 to 41%. Single-color FISH analysis revealed abundant
nuclei positive for the Y chromosome within both the
exocrine and endocrine portions of the pancreas of
control C57BL/6 male mice, whereas the pancreas of
control C57BL/6 females was devoid of the Y chromosome
(fig. S3A). We detected no evidence of engraftment, transdifferentiation,
or fusion of male splenocytes in organs including the
brain, liver, and kidney of treated NOD females (fig.
S4B), which suggests that, in addition to the low level of
hematopoietic chimerism observed, the marked incorporation of
donor cells was selective for the diseased pancreas.
To examine whether the new islet cells arose by fusion of
donor cells with endogenous islet cells, we evaluated
>800 nuclei in ß cells as well as >800 nuclei
in adjacent exocrine tissue of the five treated NOD
females studied in detail (fig. S3B and Table
2). At three scanning focal lengths, none of the regenerated
cells within the islets was enlarged compared with the
adjacent native exocrine cells. The ß-cell nuclei were
of normal size and did not contain multiple nucleoli. These observations
suggest that the regenerated islet cells were not the
products of fusion between donor splenocytes and endogenous dying
or injured ß cells, since hybrid cells contain markedly
enlarged nuclei and multiple nucleoli and are tetraploid (30,
31).
We cannot, however, exclude the possible occurrence of
fusion followed by rapid and complete expulsion of host
chromosomes.
|
|||||||||||||||||||||||||||||||||||||
We further examined the ploidy of the sex chromosomes of cells
in the regenerated islets of successfully treated NOD mice
by two-color FISH analysis with a Y
chromosome–specific probe linked to fluorescein
isothiocyanate (FITC) (green) and an X chromosome–specific
probe conjugated with cyanine 3 (Cy3) (red). A NOD
female treated with live male splenocytes exhibited only
rare if any islet cells with an apparent XXY or XXXY genotype
(Fig.
3). A normal complement of sex chromosomes was also observed
in pancreatic duct epithelial cells. These results also
indicate that the regenerated islet cells were not
likely to be the result of fusion between donor male
cells and host female cells. None of the islet cell
nuclei examined in a NOD female treated with irradiated
male splenocytes contained a detectable Y chromosome; rather,
each nucleus yielded two red signals, corresponding to
a genotype of XX (Fig.
3). Two-color FISH analysis of the pancreas of
untreated female and male NOD mice revealed that, although
this methodology can yield false-negative data (female nuclei
with no red signal or only one red signal), it almost never
yielded false-positive data (a green signal in the nucleus of
a female cell or two green signals within an individual male
nucleus) (fig. S4A).
 Fig.
3. Two-color FISH analysis of sex chromosomes in the
pancreas of NOD female mice successfully treated with
either live or irradiated male splenocytes. Pancreatic
sections from NOD females treated with live (A) or
irradiated (B) CByB6F1 male splenocytes
were subjected to nuclear staining with DAPI (blue) and
to FISH analysis with a Cy3-conjugated X
chromosome–specific probe (red dots) and an
FITC-conjugated Y chromosome–specific probe (green
dots). Purple represents overlap of Cy3 and DAPI signals.
Arrows indicate outline of islets. Fig.
3. Two-color FISH analysis of sex chromosomes in the
pancreas of NOD female mice successfully treated with
either live or irradiated male splenocytes. Pancreatic
sections from NOD females treated with live (A) or
irradiated (B) CByB6F1 male splenocytes
were subjected to nuclear staining with DAPI (blue) and
to FISH analysis with a Cy3-conjugated X
chromosome–specific probe (red dots) and an
FITC-conjugated Y chromosome–specific probe (green
dots). Purple represents overlap of Cy3 and DAPI signals.
Arrows indicate outline of islets. |
Embryonic mesenchymal cells are able to differentiate into
endothelial and endoderm cells, and they lack surface
expression of CD45 (33–37).
To examine whether nonlymphoid stem cells contribute to
the regeneration of pancreatic islets in NOD mice, we injected
12-week-old NOD females with live CD45+, CD45–,
or unfractionated CByB6F1 splenocytes
expressing enhanced green fluorescent protein (GFP).
These experiments differed from our previous experiments: (i)
The NOD females were prediabetic (with residual islet function
but with active autoimmunity) at the start of treatment;
(ii) they did not receive an islet graft; (iii) the
number of cells injected was reduced to 4 x 105
to 5 x 105 administered four times over 2
weeks; and (iv) the regrowth of islets was monitored by
detection of GFP immunofluorescence. All of the NOD females that
received CD45+, CD45–, or
unfractionated splenocytes remained normoglycemic
during the monitoring period, whereas all untreated
NOD littermates (n = 10) became diabetic.
Immunoblot analysis of cytoplasmic extracts prepared from the
pancreas of NOD mice revealed more GFP for those treated
>120 days earlier with CD45–
splenocytes than for those treated with CD45+
splenocytes (Fig.
4A). In addition, the pancreas of the prediabetic
NOD females treated with either CD45– or
unfractionated splenocytes contained islets positive for the
GFP marker (Fig.
4B). Furthermore, the newly generated islets lacked
invasive lymphocytes and were associated with minimal or
no periinsulitis, as revealed by costaining for insulin and CD45
(Fig.
4C). The proportion of islets containing cells of donor
origin was markedly smaller for prediabetic NOD hosts treated
with CD45– or unfractionated splenocytes than
for severely diabetic NOD females treated with
unfractionated splenocytes. This was as expected
because the pancreas of the prediabetic mice still
contained endogenous islets affected by
peri-insulitis. Treatment of prediabetic animals with precursor
cells thus rescues damaged islets and also promotes de novo
islet regeneration. The islets of prediabetic NOD females
treated with CD45+ splenocytes were
negative for the expression of GFP (Fig.
4B). Moreover, similar to the islet regeneration observed
in severely diabetic NOD mice treated with irradiated
splenocytes, the newly appearing islets in prediabetic
NOD females treated with CD45+ splenocytes
were devoid of invasive insulitis but exhibited
pronounced periinsulitis (Fig.
4C). The donor CD45+ splenocytes, although
essential for disease reversal, do not include cells
able to participate directly in islet generation.
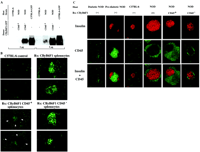 Fig.
4. Effect of treatment of prediabetic NOD mice with
CD45– or CD45+ CByB6F1
splenocytes expressing GFP on pancreatic histology. (A)
Immunoblot analysis with antibodies to GFP of pancreatic
extracts (2 or 5 µg of protein) prepared from the
indicated control mice or treated NOD mice. (B)
Prediabetic NOD females (12 weeks old) were treated with
CFA and either CD45– (lower right), CD45+
(lower left), or unfractionated (upper right) CByB6F1
splenocytes expressing GFP and were monitored for >120
days. Serial pancreatic sections containing islets
identified by costaining for insulin and CD45 were then
subjected to immunofluorescence analysis with antibodies
to GFP (green). Sections from the pancreas of an
untreated C57BL/6 mouse are shown as a control (upper
left). Arrowheads indicate peri-insulitis. (C)
Serial pancreatic sections from a diabetic NOD female, a
prediabetic NOD female (12 weeks old), and a C57BL/6
control, as well as from prediabetic NOD females treated
with CFA and either unfractionated, CD45+, or
CD45– CByB6F1 splenocytes were
subjected to immunofluorescence analysis with antibodies
to insulin (red) or to CD45 (green), as indicated; merged
images are shown in the bottom row. Fig.
4. Effect of treatment of prediabetic NOD mice with
CD45– or CD45+ CByB6F1
splenocytes expressing GFP on pancreatic histology. (A)
Immunoblot analysis with antibodies to GFP of pancreatic
extracts (2 or 5 µg of protein) prepared from the
indicated control mice or treated NOD mice. (B)
Prediabetic NOD females (12 weeks old) were treated with
CFA and either CD45– (lower right), CD45+
(lower left), or unfractionated (upper right) CByB6F1
splenocytes expressing GFP and were monitored for >120
days. Serial pancreatic sections containing islets
identified by costaining for insulin and CD45 were then
subjected to immunofluorescence analysis with antibodies
to GFP (green). Sections from the pancreas of an
untreated C57BL/6 mouse are shown as a control (upper
left). Arrowheads indicate peri-insulitis. (C)
Serial pancreatic sections from a diabetic NOD female, a
prediabetic NOD female (12 weeks old), and a C57BL/6
control, as well as from prediabetic NOD females treated
with CFA and either unfractionated, CD45+, or
CD45– CByB6F1 splenocytes were
subjected to immunofluorescence analysis with antibodies
to insulin (red) or to CD45 (green), as indicated; merged
images are shown in the bottom row. |
Overall, our data indicate that live male splenocytes injected
into female diabetic NOD mice can provide cells (CD45–
mesenchymal precursor cells) for the reconstitution of
functional islets. The donor splenocytes also
contribute to reversal of autoimmunity, possibly by
reeducating naïve T cells through presentation of
matched MHC class I molecules and self antigens, yielding
islets almost free of any signs of autoimmunity. In contrast,
diabetic NOD mice treated with irradiated splenocytes exhibit
long-term restoration of normoglycemia as a result of islet
regeneration but with markedly slower kinetics than those apparent
in NOD animals treated with live splenocytes. Thus, adult
diabetic NOD mice contain endogenous precursor cells capable
of giving rise to new islet structures after the underlying
autoimmune disease is eliminated. These syngeneic islets
appear to function normally but succumb to stable,
nonprogressive peri-insulitis.
Our findings with the NOD mouse may have implications for
treatment of diabetes or other autoimmune diseases in
humans. Both the ability of an exogenous population of
adult spleen cells to correct established diabetes
permanently and the presence of an endogenous
population of NOD mouse stem cells able to give rise
to new islets suggest that therapies to reverse autoimmune diabetes
need not incorporate transplantation of exogenous adult islets.
The use of fresh splenocytes eliminates the need for cell
culture manipulations that transform stem cells of fetal or
adult origin into malignant precursors or fusion hybrids with
an abnormal DNA content. Because the cell donors and hosts are
adults this system would preclude ethical issues associated with
the use of embryonic stem cells, as well as concerns that the
transdifferentiation of embryonic stem cells may be incomplete.
References and Notes
| D.
Faustman et al., Science 254, 1756
(1991).[ISI][Medline] |
|
| G.
Yan, Y. Fu, D. L. Faustman, J. Immunol. 159,
3068 (1997).[Abstract] |
|
| A.
A. Beg, D. |
|
4. |
T.
Hayashi, D. Faustman, Mol. Cell. Biol. 19,
8646 (1999). |
| N.
Holler et al., Nature Immunol. 1,
489 (2000).[CrossRef][ISI][Medline] |
|
| T.
Hayashi, S. Kodama, D. L. Faustman, Nature Med. 6,
1065 (2000).[CrossRef][ISI][Medline] |
|
| S.
Ryu, S. Kodama, K. Ryu, D. A. Schoenfeld, D. L. Faustman,
J. Clin. Invest. 108, 63 (2001). |
|
| M.
R. Alison et al., Nature 406, 257
(2000).[CrossRef][ISI][Medline] |
|
9. |
K.
Matsumoto, H. Yoshitomi, J. Rossant, K. S. Zaret, Science
294, 559 (2001). |
10. |
E.
Lagasse et al., Nature Med. 6, 1229
(2000).[CrossRef][ISI][Medline] |
11. |
N.
D. Theise et al., Hepatology 31, 235
(2000).[ISI][Medline] |
12. |
B.
E. Petersen et al., Science 284,
1168 (1999). |
13. |
C.
R. Bjornson, R. L. Rietze, B. A. Reynolds, M. C. Magli,
A. L. Vescovi, Science 283, 534 (1999). |
14. |
E.
Mezey, K. J. Chandross, G. Harta, R. A. Maki, S. R.
McKercher, Science 290, 1779 (2000). |
15. |
T.
R. Brazelton, F. M. V. Rossi, G. I. Keshet, H. M. Blau, Science
290, 1775 (2000). |
16. |
D.
L. Clarke et al., Science 288, 1660
(2000). |
17. |
M.
A. Eglitis, |
18. |
G.
Ferrari et al., Science 279, 1528
(1998). |
19. |
E.
Gussoni et al., Nature 401, 390
(1999).[CrossRef][ISI][Medline] |
20. |
M.
Sata et al., Nature Med. 8, 403
(2002).[CrossRef][ISI][Medline] |
21. |
F.
Quaini et al., N. Engl. J. Med. 346,
5 (2002). |
22. |
A.
A. Kocher et al., Nature Med. 7, 430
(2001).[CrossRef][ISI][Medline] |
23. |
K.
A. Jackson et al., J. Clin. Invest. 107,
1395 (2001). |
24. |
K.
R. Boheler et al., Circ. Res. 91,
189 (2002). |
| D.
Orlic et al., Ann. N.Y. Acad. Sci. 938,
221 (2001) [see discussion, p. 229]. |
|
| A.
J. Wagers, R. I. Sherwood, J. L. Christensen, |
|
| R.
F. Castro et al., Science 297, 1299 (2002). |
|
| X.
Wang et al., Nature 422, 897 (2003).[CrossRef][ISI][Medline] |
|
| G.
Vassilopoulos, P. R. Wang, D. W. Russell, Nature 422,
901 (2003).[CrossRef][ISI][Medline] |
|
| N.
Terada et al., Nature 416, 542
(2002).[CrossRef][ISI][Medline] |
|
| Q.
L. Ying, J. Nichols, E. P. Evans, A. G. Smith, Nature
416, 545 (2002).[CrossRef][ISI][Medline] |
|
| Materials
and Methods are available as supporting material at Science
Online. |
|
| Y.
Jiang et al., Nature 418, 41 (2002).[CrossRef][ISI][Medline] |
|
34. |
M.
Reyes et al., Blood 98, 2615 (2001). |
35. |
M.
Reyes et al., J. Clin. Invest. 109,
337 (2002). |
36. |
R.
E. Schwartz et al., J. Clin. Invest. 109,
1291 (2002). |
| M.
F. Pittenger et al., Science 284, 143
(1999). |
|
38. |
Supported
by the Iacocca Foundation, NIH (NIDDK grant P30
DK57521-02), the Cure Diabetes Now Foundation, and the
American Autoimmune-Related Diseases Association
Foundation. We thank J. Avruch and D. Nathan for helpful
discussions and review of the manuscript. |
Supporting Online Material
www.sciencemag.org/cgi/content/full/302/5648/1223/DC1
Materials and Methods
SOM Text
Figs. S1 to S4
Tables S1 to S3
References
8 July 2003; accepted 10 October 2003
10.1126/science.1088949
Include this information when citing this paper.
|
Volume 302, Number 5648, Issue of 14 Nov 2003, pp. 1223-1227.
 |
|
|
J Clin Invest, July 2001, Volume 108, Number
1, 63-72
Copyright ©2001 by the American Society for Clinical
Investigation
Article
|
1 Immunobiology Laboratory, and
2 Department of Biostatistics, Harvard Medical School
and Massachusetts General Hospital, Charlestown, Massachusetts,
USA
Address correspondence to: Denise L. Faustman,
Received for publication January 24, 2001, and accepted in
revised form May 14, 2001.
|
Abstract |
In NOD (nonobese diabetic) mice, a model of autoimmune diabetes,
various immunomodulatory interventions prevent progression
to diabetes. However, after hyperglycemia is
established, such interventions rarely alter the
course of disease or allow sustained engraftment of
islet transplants. A proteasome defect in lymphoid cells
of NOD mice impairs the presentation of self antigens and
increases the susceptibility of these cells to TNF-![]() –induced apoptosis.
Here, we examine the hypothesis that induction of TNF-
–induced apoptosis.
Here, we examine the hypothesis that induction of TNF-![]() expression combined with
reeducation of newly emerging T cells with self
antigens can interrupt autoimmunity. Hyperglycemic NOD
mice were treated with CFA to induce TNF-
expression combined with
reeducation of newly emerging T cells with self
antigens can interrupt autoimmunity. Hyperglycemic NOD
mice were treated with CFA to induce TNF-![]() expression and were
exposed to functional complexes of MHC class I molecules and
antigenic peptides either by repeated injection of MHC class
I matched splenocytes or by transplantation of islets from
nonautoimmune donors. Hyperglycemia was controlled in
animals injected with splenocytes by administration of
insulin or, more effectively, by implantation of
encapsulated islets. These interventions reversed the
established ß cell–directed autoimmunity and
restored endogenous pancreatic islet function to such an extent
that normoglycemia was maintained in up to 75% of animals after
discontinuation of treatment and removal of islet transplants.
A therapy aimed at the selective elimination of
autoreactive cells and the reeducation of T cells,
when combined with control of glycemia, is thus able
to effect an apparent cure of established type 1
diabetes in the NOD mouse.
expression and were
exposed to functional complexes of MHC class I molecules and
antigenic peptides either by repeated injection of MHC class
I matched splenocytes or by transplantation of islets from
nonautoimmune donors. Hyperglycemia was controlled in
animals injected with splenocytes by administration of
insulin or, more effectively, by implantation of
encapsulated islets. These interventions reversed the
established ß cell–directed autoimmunity and
restored endogenous pancreatic islet function to such an extent
that normoglycemia was maintained in up to 75% of animals after
discontinuation of treatment and removal of islet transplants.
A therapy aimed at the selective elimination of
autoreactive cells and the reeducation of T cells,
when combined with control of glycemia, is thus able
to effect an apparent cure of established type 1
diabetes in the NOD mouse.
|
Introduction |
Autoimmune destruction of pancreatic ß cells is more than
90% complete by the time hyperglycemia becomes clinically evident
in individuals with type 1 (insulin-dependent) diabetes mellitus.
Prevention of this disease would therefore optimally require
arrest of autoimmunity in the prehyperglycemic phase. After
hyperglycemia is established, therapies based on islet cell
replacement are necessary to restore physiological control of
blood glucose. Although islet transplantation has been successful
in mice, rats, and nonhuman primates with chemically
induced diabetes, sustained survival of allogeneic
islet grafts is infrequently observed in spontaneous
diabetic hosts such as the NOD (nonobese diabetic)
mouse, BB (BioBreeding) rat, and diabetic humans (1-5).
Thus, allogeneic or xenogeneic cellular grafts subjected to
transient ablation of donor MHC class I antigen expression
(6)
— achieved either with the use of "masking"
Ab’s to MHC class I molecules or by deletion of
the ß2-microglobulin (ß2M)
gene — are capable of permanent engraftment in
nonautoimmune recipients, but are minimally protected from recurrent
ß cell autoimmunity in NOD mice (7,
8).
The immune mechanisms of islet graft rejection and
recurrent autoimmunity appear distinct, and protective
interventions targeted at these two pathways of islet
destruction are nonoverlapping in effectiveness.
Subsets of antigen-presenting cells and T cells of NOD mice
that progress to hyperglycemia exhibit a decrease in the
production of LMP2, a catalytic subunit of the
proteasome, after about 5–6 weeks of age (9,
10).
This defect is accompanied by deficient generation of
peptides from endogenous proteins for display on the
cell surface by MHC class I molecules, a process that
is important for T cell memory and tolerance to self antigens
(11,
12)
and is impaired in various human and murine autoimmune diseases
(11,
13,
14).
The proteasome also contributes to the processing and
activation of NF-![]() B (15-17),
a transcription factor that regulates the expression
of genes that contribute to cytokine generation,
lymphocyte maturation, protection from TNF-
B (15-17),
a transcription factor that regulates the expression
of genes that contribute to cytokine generation,
lymphocyte maturation, protection from TNF-![]() –induced apoptosis,
and promotion of the processing of antigens for presentation
by MHC class I molecules (18-20).
The proteasome defect in NOD mice affects lymphoid
maturation as a result, at least in part, of impaired
activation of NF-
–induced apoptosis,
and promotion of the processing of antigens for presentation
by MHC class I molecules (18-20).
The proteasome defect in NOD mice affects lymphoid
maturation as a result, at least in part, of impaired
activation of NF-![]() B.
B.
In normal humans and other mammals, the continuous expression
of MHC class I molecules by peripheral cells, including
islet cells (21),
maintains peripheral tolerance in the context of properly
selected lymphocytes (12,
22).
Interruption of exposure to complexes of self peptides
and MHC class I molecules results in the aberrant
selection of CD8+ T cells that exhibit an increased
sensitivity to apoptosis (23).
Diabetic humans and NOD mice that progress to diabetes
thus manifest a paucity of memory cells and an
increased susceptibility of T cells to apoptosis, traits
that may be secondary, in part, to improper presentation of
self antigens by MHC class I molecules and ineffective T cell
selection (10,
11).
On the basis of the view that islet transplantation into
hyperglycemic NOD mice will require both the
prevention of transplant rejection and the elimination
of autoimmune T cells, we sought to combine two
strategies to achieve these goals. To bypass graft rejection,
we used donor islets from C57BL/6 mice in which the ß2M
gene was deleted. MHC class I proteins are re-expressed on
graft cells within 24 to 72 hours after
transplantation as a result of reconstitution with
host ß2M present in plasma (6,
24,
25).
The re-expression of donor MHC class I antigens is
important because it is necessary for the development and maintenance
of peripheral tolerance.
With regard to interruption of autoimmunity, we hypothesized
that the lineage-specific defects both in peptide
presentation by MHC class I molecules and in the
processing and activation of NF-![]() B might be important in the
pathogenesis of diabetes in NOD mice. The NF-
B might be important in the
pathogenesis of diabetes in NOD mice. The NF-![]() B defect in the affected
lineages on NOD mice is accompanied by an increased
sensitivity of these cells to TNF-
B defect in the affected
lineages on NOD mice is accompanied by an increased
sensitivity of these cells to TNF-![]() –induced apoptosis in
vitro (10).
The increased susceptibility to apoptosis of
misselected T cells that result from improper education
by MHC class I peptide complexes suggested that the production
of TNF-
–induced apoptosis in
vitro (10).
The increased susceptibility to apoptosis of
misselected T cells that result from improper education
by MHC class I peptide complexes suggested that the production
of TNF-![]() in
vivo might promote the selective death of such poorly
educated lineages (10,
26,
27).
Furthermore, treatment of diabetic NOD mice or BB rats
with CFA, an inducer of TNF-
in
vivo might promote the selective death of such poorly
educated lineages (10,
26,
27).
Furthermore, treatment of diabetic NOD mice or BB rats
with CFA, an inducer of TNF-![]() production, both impairs the
transfer of disease by T cells from these animals to
naive hosts (28-31)
as well as prolongs the survival of syngeneic islet
grafts in spontaneously diabetic hosts (2,
32).
We therefore hypothesized that CFA treatment might
eliminate, at least temporarily, the autoreactive lymphoid cells
of NOD mice by promoting their apoptosis, in part through the
induction of TNF-
production, both impairs the
transfer of disease by T cells from these animals to
naive hosts (28-31)
as well as prolongs the survival of syngeneic islet
grafts in spontaneously diabetic hosts (2,
32).
We therefore hypothesized that CFA treatment might
eliminate, at least temporarily, the autoreactive lymphoid cells
of NOD mice by promoting their apoptosis, in part through the
induction of TNF-![]() .
.
Thus, we both treated hyperglycemic NOD recipients with CFA,
seeking to eliminate autoreactive lymphoid lineages, and
transplanted islets from ß2M-deficient
donors under the kidney capsule of these animals,
seeking to generate graft-specific tolerance. These
interventions resulted in the marked reduction or
apparent elimination of ongoing ß cell–directed autoimmunity.
Unexpectedly, the long-term reappearance of endogenous ß
cell function was also observed in the pancreatic islets
of the previously hyperglycemic hosts.
|
Methods |
Animals. Female NOD mice from Taconic Farms
(Germantown, New York, USA) and C57BL/6J (C57) mice
from The Jackson Laboratory (Bar Harbor, Maine, USA)
were maintained under pathogen-free conditions. NOD
mice were screened for the onset of diabetes by monitoring body
weight and blood glucose; they were diagnosed as diabetic when
two consecutive blood glucose concentrations exceeded 400 mg/dl.
Before experimental treatments, diabetic NOD mice were maintained
for 7–20 days on daily injections of 1.0–1.5 U
of NPH human insulin per 100 g of body weight, thereby preventing
immediate death and maintaining blood sugar concentration
between 200 and 700 mg/dl. The use of such severely
diabetic mice, relatively late after disease onset,
ensured that endogenous pancreatic islets were
completely obliterated before the initiation of experiments.
Splenocyte donors included normal C57 mice, C57 mice
(C57 ß2M–/–)
in which the ß2M gene was disrupted, C57
mice (C57 ß2M–/–,
TAP1–/–) in which both
the ß2M and Tap1 genes were
disrupted, and MHC class II–/–
mice (C57 class II–/–)
in which the I-A gene was disrupted and the E locus of
MHC class II was not expressed because of an
endogenous defect in the C57 strain (Taconic Farms). Splenocytes
(9 x 106) were injected into NOD recipients
through the tail vein twice a week. CFA (Difco
Laboratories, Detroit, Michigan, USA) was freshly
mixed with an equal volume of physiological saline and
injected (50 µl) into each hind-foot pad at the time
of islet transplantation or after the first splenocyte injection.
Islet transplantation. Islets were isolated
from donor C57 mice or 6- to 8-week-old prediabetic
female NOD mice. Density gradient centrifugation followed
by hand picking of islets ensured that both preparations were
highly enriched in islets and had an accurate determination of
islet number. For transplantation, 500–600 freshly isolated
islets were grafted beneath the left renal capsule of
each diabetic NOD recipient. For islet encapsulation,
900–1,100 islets were enclosed in alginate
spheres, which were then surgically inserted into the
peritoneal cavity of diabetic NOD mice. Transplantation was
considered successful if the nonfasting blood concentration of
glucose returned to normal (<200 mg/dl) within 24 hours after
surgery. The glucose concentration of orbital blood was monitored
three times a week after transplantation with a Glucometer Elite
instrument (Bayer Corp, Pittsburgh, Pennsylvania, USA). Body
weight was also monitored three times a week. Islet grafts were
considered to have been rejected if the blood glucose
concentration increased to more than 250 mg/dl on two
occasions. Recipients that rejected islet grafts were
killed for histological examination and
flow-cytometric studies. To assess the contribution of endogenous
pancreatic islets to the control of blood sugar
concentration, we removed subrenal islet transplants
by nephrectomy. Similarly, islets encapsulated in
alginate spheres, which were approximately 0.2–0.5
cm in diameter, were removed from the peritoneal cavity
by direct visualization under a dissecting microscope. Histological
analysis of the pancreata and islet grafts was performed
by staining with hematoxylin and eosin for evaluation of
lymphocytic infiltrates and with aldehyde-fuchsin for islet insulin
content. The entire pancreas from splenic to duodenal stomach
attachments was removed, fixed, embedded longitudinally in
a paraffin block, and subjected to serial sectioning (10 µm).
Flow cytometry. Spleens were removed and
gently minced on a stainless steel sieve. Cell
suspensions were rendered free of red blood cells by
exposure to a solution containing 0.83% NH4Cl. The splenocytes
were stained with mouse mAb’s (PharMingen, San Diego,
California, USA) to CD8 (FITC-labeled), to CD62L
(phycoerythrin-labeled [PE-labeled]), to CD95
(PE-labeled), or to CD45RB (PE-labeled), and were
analyzed (>10,000 cells per sample) the same day with
an Epics Elite flow cytometer. Spleen cells were incubated for
24 hours in the absence or presence of TNF-![]() (20 ng/ml), after
which apoptotic cells were detected by flow cytometry with
FITC-conjugated annexin V. Apoptotic cells were defined as
cells positive for both propidium iodide (PI) and annexin V
staining; numbers within the upper quadrants represent the corresponding
percentages of cells.
(20 ng/ml), after
which apoptotic cells were detected by flow cytometry with
FITC-conjugated annexin V. Apoptotic cells were defined as
cells positive for both propidium iodide (PI) and annexin V
staining; numbers within the upper quadrants represent the corresponding
percentages of cells.
Adoptive transfer. Adoptive transfer was
performed as described (29).
Recipient male NOD mice, 4–8 weeks of age, were
irradiated (790 rads) with a 137Cs source
and injected intravenously within 2 hours of
irradiation with donor splenocytes (2 x 107 viable
cells) suspended in 0.25 ml of serum-free medium. Diabetic
spleen cell donors were female NOD mice that typically
had exhibited blood sugar concentrations of greater
than 400 mg/dl for at least 3 weeks. Multiple diabetic
donor spleens were pooled to produce sufficient cells
for all of the hosts in a given experiment.
Statistics. Exact algorithm P values
were calculated in some instances with multiple
comparisons corrected by the number of tested variables. A
P value less than 0.05 was considered statistically
significant.
|
Results |
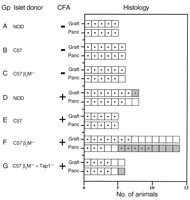
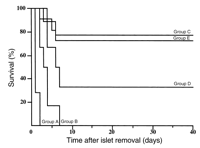
CFA treatment and islet transplantation in NOD mice.
Hosts for the transplantation experiments were severely diabetic
female NOD mice, usually more than 20 weeks of age, that
had exhibited blood glucose concentrations of greater
than 400 mg/dl for at least 7 days and had been
treated by daily administration of insulin to prevent
death. Islet transplants were placed unilaterally under
the kidney capsule to facilitate nonlethal removal and histological
examination. Islets from 6- to 8-week-old prediabetic NOD
females (recipient group A) or from normal C57 mice (recipient
group B) were rapidly rejected by diabetic NOD recipients
(Table 1,
Figure 1a).
Although C57 donor islets with transient ablation of
MHC class I expression survive indefinitely in nonautoimmune
diabetic hosts (6),
the survival time of islets from ß2M–/–
C57 mice in diabetic NOD females (group C) was only about
three times that of normal C57 islets. As expected
from previous observations (2,
32),
treatment with CFA prolonged the survival of syngeneic islet
grafts in diabetic NOD hosts (group D) but had a minimal effect
on the survival of C57 islets (group E), which were uniformly
rejected by 11 days after transplantation. However, the
combination of ß2M–/–
C57 islet transplants with CFA treatment resulted in
sustained (>129 days) normoglycemia in 5 of 14
diabetic NOD hosts (group F). Although the duration of
hyperglycemia before initiation of therapy varied between 7
and 20 days, the length of this interval was not significantly
related to the duration of sustained normoglycemia after
treatment (data not shown). The animals that exhibited
sustained normoglycemia also demonstrated progressive
weight gain, similar to that apparent in NOD female
cohorts who never became diabetic (data not shown). With
normalization of blood sugar concentration as a measure of
treatment success, the success of ß2M–/–
C57 islet transplantation together with CFA administration
was significantly different from that apparent with
the other groups combined but did not differ from that
of NOD islet transplantation together with CFA
treatment.
|
|
After the recurrence of hyperglycemia in the NOD mice that had
been treated with CFA and syngeneic (NOD) islet
transplants, the kidney containing the islet graft was
examined histologically. Marked lymphocytic
infiltration was apparent under the kidney capsule at
the site of transplantation, a characteristic of recurrent
autoimmune disease (Figure 1b,
group D); moreover, no intact islets were detected in
the pancreas, although islet remnants, largely
obscured by dense pockets of infiltrating lymphocytes,
were evident. Similar histological characteristics of
both the transplant site and pancreas were apparent in diabetic
NOD mice that had received CFA and islet grafts from C57
donors (Figure 1b,
group E). Unexpectedly, for all five NOD mice with long-term
normoglycemia after receiving ß2M–/–
C57 islets and CFA treatment, no surviving islet grafts
were detected under the kidney capsule when the
animals were killed at more than 129 days after
transplantation (Figure 1b,
group F). In contrast, the pancreas of each of these
five recipients exhibited well-formed islets that
appeared completely granulated when stained by
aldehyde-fuchsin. The islets were free of lymphocytes or
lymphocytes were present only circumferentially; this latter
pattern of lymphocyte accumulation, with lymphocytes
surrounding but not invading the islets, has been
associated with nonprogressive or interrupted ß cell
autoimmunity (33).
The return to normoglycemia in the absence of
detectable transplanted islet tissue, together with
the presence of islets in a pancreas largely devoid of
infiltrating lymphocytes, suggested not only that autoimmunity
had been interrupted but that the function of endogenous ß
cells had been restored.
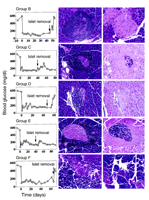
Restoration of near-normal pancreatic islet histology was
observed only in the diabetic NOD mice that received
both ß2M–/–
islet grafts and CFA treatment (Figure 2).
Pancreatic islets were thus not detected in any
diabetic NOD mice treated with CFA and syngeneic NOD
islets; the persistence of normoglycemia in such
recipients appeared solely due to the transplanted islets, which
always exhibited invasive insulitis (Figure 1b,
Figure 2).
Thus, treatment with CFA together with syngeneic NOD islets may
have slowed disease recurrence, but persistent autoimmunity remained.
|
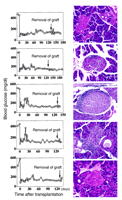
The relative contributions of restored endogenous pancreatic
islets and transplanted islets to the maintenance of
normoglycemia in NOD mice treated with CFA and islet
grafts from ß2M–/–
C57 donors were assessed by removal of the kidney
containing the islet transplant after 120 days of
normoglycemia in a second group of five animals. All
five mice remained normoglycemic after nephrectomy
until they were killed 3–60 days later (Figure 3).
Histological analysis of the kidneys that received the
grafts revealed a complete loss of identifiable islet structures.
In contrast, the pancreata of all five recipients contained
well-formed islets either without lymphocytic infiltration
or with circumferentially distributed lymphocytes
only. Normoglycemia after nephrectomy was thus
maintained solely by endogenous pancreatic islets. In
contrast, nephrectomies performed during the posttransplantation
period of normoglycemia (day 62, day 85) in two mice who
had received CFA plus syngeneic NOD islets resulted in
a rapid return to hyperglycemia (data not shown),
demonstrating that the control of blood sugar in this
treatment group was mediated solely by the
transplanted islet tissue.
|
We also transplanted diabetic NOD females with islets from C57
mice in which the genes for both ß2M and TAP1
had been deleted. Together with TAP2, TAP1 mediates
the transport of endogenous peptides from the cytosol
into the lumen of the endoplasmic reticulum for their
assembly with MHC class I molecules (34).
Islet cells from these mice are more permanently defective in
presentation of self antigens by MHC class I than are those from
ß2M–/–
mice. Transplantation of ß2M–/–,
TAP1–/– C57 islets
combined with injection of CFA resulted in the return
of hyperglycemia within 14 days in five of six animals
(group G); histological examination of the pancreata revealed
a pattern typical of that for untreated diabetic NOD mice
(Table 1,
Figure 2).
Thus, a transient interruption of peptide presentation
by donor MHC class I molecules is essential for the
abrogation of autoimmunity, whereas a sustained interruption
of this process prevents the reestablishment of tolerance
and the restoration of endogenous pancreatic islet
integrity.
CFA treatment and splenocyte injection in NOD mice.
Given that the restoration of normoglycemia in the diabetic NOD
mice treated with CFA and ß2M–/–
C57 islets did not depend on the continuing secretion
of insulin by the islet grafts, we next investigated
whether C57 donor cell types other than islets might
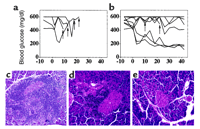
serve a similar therapeutic role. Nine diabetic NOD
mice were treated with a single bilateral injection of
CFA followed by a 40-day regimen of biweekly intravenous injections
of C57 splenocytes. These lymphoid cells express both
MHC class I and class II proteins and survive only transiently
in NOD hosts because of graft rejection (data not shown).
Repeat injections of splenocytes ensure that the host
is continuously exposed to intact antigen presentation
complexes on the surface of these cells. The
recipients were monitored for hyperglycemia every 3 or
4 days, and insulin was administered daily unless normoglycemia
returned. A control group of four diabetic NOD mice
received daily insulin injections only. All four control mice
died on or before day 25 of the experimental period as a
result of poor control of blood glucose and consequent ketosis
and cachexia (Figure 4a).
In contrast, seven of the nine mice injected with CFA
and C57 splenocytes were alive after 40 days, and
three of these animals had become normoglycemic and insulin independent
(Figure 4b).
The pancreata of control (insulin treatment only) mice
exhibited pronounced lymphocytic infiltrates that obscured
any remaining islet structures (Figure 4c).
The pancreata of the four NOD mice treated with CFA
and C57 splenocytes that remained alive but
hyperglycemic and insulin dependent revealed a marked
decrease (relative to control animals) in the number of
lymphoid infiltrates, which were located either circumferentially
or adjacent to the infrequent islet structures (Figure 4d).
On killing of each of the three NOD mice treated with CFA
and C57 splenocytes that maintained normoglycemia
after discontinuation of insulin injections, the
pancreata exhibited abundant islets that were free of
invasive lymphocytes or were associated only with
circumferential lymphocytes (Figure 4e).
Thus, treatment with CFA combined with repeated
exposure to C57 lymphocytes resulted in complete
reversal of diabetes in approximately 30% of NOD
recipients and partially restored ß cell function in
an additional approximately 40% of recipients.
|
Influence of glycemic control on restoration of
endogenous islet function. The reversal of diabetes in
NOD mice by CFA and repeated exposure to C57
splenocytes indicated that restoration of endogenous islet
function is achievable without islet transplantation and despite
the poor glycemic control attained by insulin injections. The
beneficial influence of glycemic control on the growth, survival,
and function of cultured islets, as well as of transplanted islets
in nonautoimmune settings, has been demonstrated (35,
36).
To determine whether the restoration of endogenous ß cell
function could be achieved more consistently with better control
of blood glucose, we replaced insulin injections with the
intraperitoneal implantation of alginate-encapsulated C57 mouse
islets. Alginate encapsulation prevents direct contact between
donor endocrine cells and host T cells, and such grafts have
been shown to provide near-normal glycemic control for 40
to 50 days in approximately 70–80% of autoimmune NOD recipients
(37).
Almost all diabetic NOD mice that received
alginate-encapsulated C57 islets exhibited improved
glucose regulation or normoglycemia. The alginate
spheres were removed 40–50 days after implantation, and
blood glucose concentration was monitored (Table 2).
The seven mice treated only with alginate-encapsulated
islets (group A), the six mice that received a single
bilateral injection of CFA (group B), and the three
mice treated with biweekly injections of C57
splenocytes (data not shown) all exhibited a rapid return to
hyperglycemia and early death after removal of the implants (Table
2,
Figure 5).
The pancreata of NOD mice that received only
alginate-encapsulated islets revealed no sign of intact islets
or of lymphoid infiltrates (data not shown). The pancreata of
NOD hosts treated with CFA and alginate-encapsulated islets exhibited
marked invasive insulitis that obscured islet structures (Figure
6).
In contrast, seven of the nine (78%) diabetic NOD mice
that received CFA and C57 splenocytes (group C) remained normoglycemic
for more than 40 days (until killing) after removal of
the alginate-encapsulated islets (Figure 5,
Table 2).
The pancreata of these animals contained large islets
with circumferentially distributed lymphocytes (Figure
6).
The islet mass after at least 80 days of disease
reversal was estimated at approximately 50% of the
original value. The pancreata from control BALB/c mice
contained approximately 25–35 islets; the pancreata from
successfully treated NOD mice contained approximately 12–20
islets with serial histological sections. Thus, maintenance
of normoglycemia during the treatment period increased the
percentage of diabetic mice cured of hyperglycemia.
|
|
|
Role of TNF-![]() in treatment
outcome. We attempted to identify features of the
successful treatment regimens that are critical to a
positive outcome. We had used CFA to induce the
endogenous production of TNF-
in treatment
outcome. We attempted to identify features of the
successful treatment regimens that are critical to a
positive outcome. We had used CFA to induce the
endogenous production of TNF-![]() (31).
The importance of TNF-
(31).
The importance of TNF-![]() in treatment success was
therefore investigated by the intravenous
administration of a rat IgG1 mAb to this cytokine (clone
MP6-X73; Accurate Chemical & Scientific Corp., Westbury,
New York, USA) at a dose of 1.5 mg/day for the first 10
days in diabetic NOD hosts treated with C57
splenocytes, CFA, and alginate-encapsulated islets.
All five NOD mice so treated exhibited a rapid return
to hyperglycemia on removal of the alginate-encapsulated islets
50–70 days after transplantation (Table 2,
group F), consistent with the notion that TNF-
in treatment success was
therefore investigated by the intravenous
administration of a rat IgG1 mAb to this cytokine (clone
MP6-X73; Accurate Chemical & Scientific Corp., Westbury,
New York, USA) at a dose of 1.5 mg/day for the first 10
days in diabetic NOD hosts treated with C57
splenocytes, CFA, and alginate-encapsulated islets.
All five NOD mice so treated exhibited a rapid return
to hyperglycemia on removal of the alginate-encapsulated islets
50–70 days after transplantation (Table 2,
group F), consistent with the notion that TNF-![]() plays an obligatory role
in the beneficial effect of CFA. This effect of the mAb to
TNF-
plays an obligatory role
in the beneficial effect of CFA. This effect of the mAb to
TNF-![]() was
specific, given that administration of a rat IgG1 mAb
to the human T cell receptor Vß1 chain (clone BL37.2;
American Type Culture Collection, Rockville, Maryland, USA)
at a dose of 1.5 mg/day for 10 days did not affect the success
of treatment with C57 splenocytes and CFA (data not shown).
Direct administration of TNF-
was
specific, given that administration of a rat IgG1 mAb
to the human T cell receptor Vß1 chain (clone BL37.2;
American Type Culture Collection, Rockville, Maryland, USA)
at a dose of 1.5 mg/day for 10 days did not affect the success
of treatment with C57 splenocytes and CFA (data not shown).
Direct administration of TNF-![]() to diabetic hosts was not
feasible because of the prohibitive cost.
to diabetic hosts was not
feasible because of the prohibitive cost.
We next investigated whether the production of TNF-![]() in diabetic NOD
mice treated with CFA results in the selective elimination of
autoreactive lymphoid cells first by examining the susceptibility
of lymphocytes from successfully treated animals to TNF-
in diabetic NOD
mice treated with CFA results in the selective elimination of
autoreactive lymphoid cells first by examining the susceptibility
of lymphocytes from successfully treated animals to TNF-![]() –induced cell
death in vitro. As shown previously (26,
27),
incubation of normal C57 spleen cells with TNF-
–induced cell
death in vitro. As shown previously (26,
27),
incubation of normal C57 spleen cells with TNF-![]() in vitro had no effect on
cell viability; for the animal shown in Figure 7a,
the proportion of apoptotic cells was 0.01% for
splenocytes incubated in the absence or presence of TNF-
in vitro had no effect on
cell viability; for the animal shown in Figure 7a,
the proportion of apoptotic cells was 0.01% for
splenocytes incubated in the absence or presence of TNF-![]() . In contrast, exposure of
splenocytes from an untreated 20-week-old NOD female
to TNF-
. In contrast, exposure of
splenocytes from an untreated 20-week-old NOD female
to TNF-![]() in
vitro increased the proportion of apoptotic cells from
0.03 to 38.3%. Such an increased sensitivity to TNF-
in
vitro increased the proportion of apoptotic cells from
0.03 to 38.3%. Such an increased sensitivity to TNF-![]() –induced apoptosis in
vitro was no longer evident with spleen cells derived
from NOD mice cured of diabetes; thus, splenocytes
from a NOD female successfully treated with both CFA
and C57 splenocytes (Table 2,
group C) exhibited 23.2 and 23.9% apoptosis in the
absence and presence of TNF-
–induced apoptosis in
vitro was no longer evident with spleen cells derived
from NOD mice cured of diabetes; thus, splenocytes
from a NOD female successfully treated with both CFA
and C57 splenocytes (Table 2,
group C) exhibited 23.2 and 23.9% apoptosis in the
absence and presence of TNF-![]() , respectively (Figure 7a).
Successful therapy generated a subpopulation of
nonpathologic but TNF-
, respectively (Figure 7a).
Successful therapy generated a subpopulation of
nonpathologic but TNF-![]() –resistant T cells
that exhibited an increased tendency to undergo cell death in
culture (Figure 7a).
Disease reversal, even 210 days after cessation of
treatment, was thus associated with the persistent elimination
of TNF-
–resistant T cells
that exhibited an increased tendency to undergo cell death in
culture (Figure 7a).
Disease reversal, even 210 days after cessation of
treatment, was thus associated with the persistent elimination
of TNF-![]() –sensitive
T cells, a population of cells with a demonstrated
ability to play a role in disease (29,
30).
The permanent elimination of these formerly abundant TNF-
–sensitive
T cells, a population of cells with a demonstrated
ability to play a role in disease (29,
30).
The permanent elimination of these formerly abundant TNF-![]() –sensitive lymphoid
cells, presumably in response to TNF-
–sensitive lymphoid
cells, presumably in response to TNF-![]() (and, perhaps, to other
CFA-induced cytokines), was observed uniformly in
successfully treated diabetic NOD mice. Similar
complete and stable elimination of TNF-
(and, perhaps, to other
CFA-induced cytokines), was observed uniformly in
successfully treated diabetic NOD mice. Similar
complete and stable elimination of TNF-![]() –sensitive cells
at various times after treatment has been observed in more
than 20 NOD mice.
–sensitive cells
at various times after treatment has been observed in more
than 20 NOD mice.
|
We also examined the effect of TNF-![]() on the pathogenesis of
autoimmune diabetes in adoptive transfer experiments.
Young recipient NOD males were subjected to
irradiation followed by an intravenous injection of
donor splenocytes either from newly diabetic NOD females
or from NOD mice with long-term normoglycemia due to treatment
with CFA and C57 splenocytes. The onset of diabetes was
observed in all recipients by day 15 after the transfer of
diabetic mouse cells that were injected either immediately after
isolation or after control culture for 24 hours (Figure 7b,
left panel). In contrast, four of the five recipients of diabetic
mouse cells that had been cultured with TNF-
on the pathogenesis of
autoimmune diabetes in adoptive transfer experiments.
Young recipient NOD males were subjected to
irradiation followed by an intravenous injection of
donor splenocytes either from newly diabetic NOD females
or from NOD mice with long-term normoglycemia due to treatment
with CFA and C57 splenocytes. The onset of diabetes was
observed in all recipients by day 15 after the transfer of
diabetic mouse cells that were injected either immediately after
isolation or after control culture for 24 hours (Figure 7b,
left panel). In contrast, four of the five recipients of diabetic
mouse cells that had been cultured with TNF-![]() for 24 hours
before adoptive transfer remained normoglycemic for at least
40 days after cell injection (Figure 7b,
middle panel). Furthermore, the four NOD hosts each
injected with splenocytes from a different NOD female
that had experienced reversal of autoimmunity for more
than 120 days failed to develop disease during
observation periods of more than 60 days (Figure 7b,
right panel). TNF-
for 24 hours
before adoptive transfer remained normoglycemic for at least
40 days after cell injection (Figure 7b,
middle panel). Furthermore, the four NOD hosts each
injected with splenocytes from a different NOD female
that had experienced reversal of autoimmunity for more
than 120 days failed to develop disease during
observation periods of more than 60 days (Figure 7b,
right panel). TNF-![]() –resistant NOD
splenocytes, enriched either in vitro by direct
exposure of cells to TNF-
–resistant NOD
splenocytes, enriched either in vitro by direct
exposure of cells to TNF-![]() or in vivo by
disease reversal, are thus incapable of transferring disease
to naive hosts.
or in vivo by
disease reversal, are thus incapable of transferring disease
to naive hosts.
Role of MHC class I peptide complexes in T cell
selection and treatment outcome. Disease reversal in
diabetic NOD mice required treatment with both CFA and
cells that express MHC class I peptide complexes. Only
two of six (33%) diabetic NOD mice that received CFA and biweekly
injections of splenocytes from ß2M–/–,
TAP1–/– C57 donors
remained normoglycemic after removal of
alginate-encapsulated islets (Table 2,
group D). The pancreata of the four animals that
became hyperglycemic after removal of the alginate
spheres contained no granulated islets and only a few
visible islet structures, which were invaded and
obscured by lymphocytic infiltrates (Figure 6).
In contrast, 8 of 11 (73%) diabetic NOD mice treated
with CFA and splenocytes from C57 donors lacking MHC
class II protein expression remained normoglycemic
after removal of the alginate-encapsulated islets (Table
2,
group E); the pancreata of these animals contained large
islets that exhibited only moderate lymphocytic accumulation
at the periphery (Figure 6).
Interruption of antigen presentation by MHC class I skews the
T cell repertoire to an overabundance of naive cells, a
consistent trait of diabetes-prone NOD mice and humans
(38-40).
Improper T cell selection secondary to interruption of
antigen presentation by MHC class I results in
overexpression of CD95 by CD8+ T cells as
well as an increase in the abundance of cells with naive cell
markers such as CD62L+ and CD45RBhigh (12,
23).
To investigate whether therapeutic reversal of NOD
mouse diabetes was associated with a change in naive T
cell selection, we subjected splenocytes to
flow-cytometric analysis. Flow cytometry was performed 5 to
26 days after removal of the alginate-encapsulated islets and
termination of therapy.
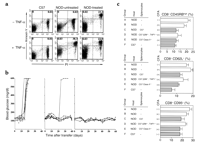
Untreated NOD mice exhibited the expected increases in the
abundance of naive CD8+CD45RBhigh,
CD8+CD62L+, and CD8+CD95+
cells compared with C57 animals (Figure 7c).
In contrast, in NOD female mice that were successfully
treated with alginate-encapsulated islets, CFA, and
administration of C57 splenocytes, the percentages of
each of these cell populations were reduced to normal or near-normal
values. The abnormally high numbers of CD8+CD45RBhigh,
CD8+CD62L+, and CD8+CD95+
cells remained increased in diabetic NOD females
treated with alginate-encapsulated islets and CFA, either
alone or together with administration of ß2M–/–,
TAP1–/– C57 splenocytes.
Data are means plus or minus SEM of values from at
least five mice per group. Exact algorithm P
values were calculated for comparisons of each cell population
between groups C, E, and F versus groups A, B, and D: P
= 0.001 for CD8+CD45RBhigh cells, P
= 0.01 for CD8+CD62L+ cells, and P =
0.05 for CD8+CD95+ cells. Despite the fact
that many comparisons were performed, the P
value remained less than 0.05 even when multiplied by
the three comparisons. These data showed that the T
cell reselection apparent in successfully treated NOD
mice was secondary to reexposure to complexes of MHC
class I molecules and self peptides. The normalization of T
cell phenotype did not require reexposure to MHC class
II–peptide complexes, given that the
administration together with CFA and alginate-encapsulated
islets of MHC class II–/– splenocytes
was as effective as was that of normal C57 splenocytes.
|
Discussion |
We have demonstrated the effectiveness of a novel therapy for
the correction of established autoimmune diabetes in the
NOD mouse. Three aspects of this treatment regimen
appear to operate in parallel and in a synergistic
manner: (a) Injection of CFA, and the consequent
induction of TNF-![]() , results in the elimination
of TNF-
, results in the elimination
of TNF-![]() –sensitive cells, which
have been shown previously to transfer existing
disease (28-31);
(b) the introduction of functional MHC class I peptide
complexes expressed on the surface of either normal
islet cells or normal lymphocytes results in partial
but stable reselection of the T cell population of the NOD
host, leading to an increase in the abundance of long-term memory
T cells (6,
12,
23);
and (c) suppression of hyperglycemia, although not
obligatory, promotes the functional restoration of
endogenous ß cells or their precursors.
–sensitive cells, which
have been shown previously to transfer existing
disease (28-31);
(b) the introduction of functional MHC class I peptide
complexes expressed on the surface of either normal
islet cells or normal lymphocytes results in partial
but stable reselection of the T cell population of the NOD
host, leading to an increase in the abundance of long-term memory
T cells (6,
12,
23);
and (c) suppression of hyperglycemia, although not
obligatory, promotes the functional restoration of
endogenous ß cells or their precursors.
We propose that continuous or repeated exposure to parenchymal
or lymphoid cells expressing MHC class I molecules and self
peptides initiates the reeducation of host T cells, which
was apparent in CFA-treated hosts from the loss of
cells with an increased sensitivity to TNF-![]() –induced apoptosis and
from the restoration of a cell surface phenotype
characteristic of long-term memory cells. This
reeducation resulted in the establishment of long-term
tolerance, as demonstrated by the elimination of both
recurrent hyperglycemia and invasive insulitis. Treatment of
NOD mice with severe hyperglycemia and islet destruction resulted
in the reappearance of pancreatic insulin-secreting cells
and normoglycemia. The rate of pancreatic ß cell
proliferation is increased during the active phase of disease
in NOD mice, and NOD islet stem cells proliferate in
culture (41).
The interruption of ß cell autoimmunity may promote
both the rescue of surviving ß cells in islets as
well as the production of new ß cells that are now
able to survive in the altered immunological milieu. The expression
of MHC class I molecules and self peptides by NOD pancreatic
ß cells (21)
may be responsible for maintenance of peripheral
tolerance after termination of disease by transient therapy.
–induced apoptosis and
from the restoration of a cell surface phenotype
characteristic of long-term memory cells. This
reeducation resulted in the establishment of long-term
tolerance, as demonstrated by the elimination of both
recurrent hyperglycemia and invasive insulitis. Treatment of
NOD mice with severe hyperglycemia and islet destruction resulted
in the reappearance of pancreatic insulin-secreting cells
and normoglycemia. The rate of pancreatic ß cell
proliferation is increased during the active phase of disease
in NOD mice, and NOD islet stem cells proliferate in
culture (41).
The interruption of ß cell autoimmunity may promote
both the rescue of surviving ß cells in islets as
well as the production of new ß cells that are now
able to survive in the altered immunological milieu. The expression
of MHC class I molecules and self peptides by NOD pancreatic
ß cells (21)
may be responsible for maintenance of peripheral
tolerance after termination of disease by transient therapy.
The application of this therapy to humans with type 1 diabetes
may be feasible. As in NOD mice, lymphocytes from type 1
diabetic humans show an increased sensitivity to TNF-![]() –induced apoptosis (10)
as well as age-related defects in MHC class I presentation of
self peptides for proper T cell selection (11,
13).
Moreover, diabetic humans continue to produce
auto-Ab’s to islet targets for several years
after the onset of frank hyperglycemia, indicating the
persistence of islet cell antigen expression. Thus, a
proportion of individuals with type 1 diabetes may possess a
ß cell mass or islet regenerative potential similar to
that of hyperglycemic NOD mice. Even if the regenerative capacity
of ß cells is exhausted, a similar immunomodulation approach
may provide a less hostile milieu for islet replacement.
–induced apoptosis (10)
as well as age-related defects in MHC class I presentation of
self peptides for proper T cell selection (11,
13).
Moreover, diabetic humans continue to produce
auto-Ab’s to islet targets for several years
after the onset of frank hyperglycemia, indicating the
persistence of islet cell antigen expression. Thus, a
proportion of individuals with type 1 diabetes may possess a
ß cell mass or islet regenerative potential similar to
that of hyperglycemic NOD mice. Even if the regenerative capacity
of ß cells is exhausted, a similar immunomodulation approach
may provide a less hostile milieu for islet replacement.
|
Acknowledgments |
This work was supported by The Iacocca Foundation. We thank Biohybrid
Technologies (J. Hayes, D. Wolf, and C. McGrath) for assistance
with islet preparation and encapsulation; S. Thompson (Bayer
Corp.) for providing surplus blood glucose monitoring strips;
M. Contant for preparation of specimens for histological analysis;
NICHD for funding to study autoimmune PDF patients; and
J. Avruch and D. Nathan for critical review of the manuscript.
|
Footnotes |
Shinichiro Ryu and Shohta Kodama contributed equally to this
work.
|
References |
This article has been cited by other articles:
|
|
S. Kodama, W. Kuhtreiber, S. Fujimura, E. A. Dale, and D. L. Faustman Islet Regeneration During the Reversal of Autoimmune Diabetes in NOD Mice Science, November 14, 2003; 302(5648): 1223 - 1227. [Abstract] [Full Text] [PDF] |
|||
|
|
G. Demirci, T. B. Strom, and X. C. Li Islet Allograft Rejection in Nonobese Diabetic Mice Involves the Common {gamma}-Chain and CD28/CD154-Dependent and -Independent Mechanisms J. Immunol., October 1, 2003; 171(7): 3878 - 3885. [Abstract] [Full Text] [PDF] |
|||
|
|
T. D. Zorina, V. M. Subbotin, S. Bertera, A. M. Alexander, C. Haluszczak, B. Gambrell, R. Bottino, A. J. Styche, and M. Trucco Recovery of the Endogenous {beta} Cell Function in the NOD Model of Autoimmune Diabetes Stem Cells, July 1, 2003; 21(4): 377 - 388. [Abstract] [Full Text] [PDF] |
|||
|
|
Y. C. Zhang, A. Pileggi, A. Agarwal, R. D. Molano, M. Powers, T. Brusko, C. Wasserfall, K. Goudy, E. Zahr, R. Poggioli, M. Scott-Jorgensen, M. Campbell-Thompson, J. M. Crawford, H. Nick, T. Flotte, T. M. Ellis, C. Ricordi, L. Inverardi, and M. A. Atkinson Adeno-Associated Virus-Mediated IL-10 Gene Therapy Inhibits Diabetes Recurrence in Syngeneic Islet Cell Transplantation of NOD Mice Diabetes, March 1, 2003; 52(3): 708 - 716. [Abstract] [Full Text] [PDF] |
|||
|
|
D. L. FAUSTMAN Reversal of Established Autoimmune Diabetes by in Situ {beta}-Cell Regeneration Ann. N.Y. Acad. Sci., June 1, 2002; 961(1): 40 - 40. [Full Text] [PDF] |
|||
|
|
A. Rabinovitch, W. L. Suarez-Pinzon, A.M. J. Shapiro, R. V. Rajotte, and R. Power Combination Therapy With Sirolimus and Interleukin-2 Prevents Spontaneous and Recurrent Autoimmune Diabetes in NOD Mice Diabetes, March 1, 2002; 51(3): 638 - 645. [Abstract] [Full Text] [PDF] |
|||
|
|
J. P. Palmer Immunomodulatory therapy of human type 1 diabetes: lessons from the mouse J. Clin. Invest., July 1, 2001; 108(1): 31 - 33. [Full Text] [PDF] |
|||
|
|
Copyright
© 2001 by the American Society for Clinical Investigation.
|
||
|
|
J Clin Invest, July 2001, Volume 108, Number
1, 63-72
Copyright ©2001 by the American Society for Clinical
Investigation
Article
|
1 Immunobiology Laboratory, and
2 Department of Biostatistics, Harvard Medical School
and Massachusetts General Hospital, Charlestown, Massachusetts,
USA
Address correspondence to: Denise L. Faustman,
Immunobiology Laboratory, Massachusetts General
Hospital–East, Harvard Medical School, Building 149, Room
3602, 13th Street, MailStop M1493601, Charlestown, Massachusetts
02129, USA. Phone: (617) 726-4084; Fax: (617) 726-4095; E-mail: denise.faustman@cbrc2.mgh.harvard.edu.
Received for publication January 24, 2001, and accepted in
revised form May 14, 2001.
|
Abstract |
In NOD (nonobese diabetic) mice, a model of autoimmune diabetes,
various immunomodulatory interventions prevent progression
to diabetes. However, after hyperglycemia is
established, such interventions rarely alter the
course of disease or allow sustained engraftment of
islet transplants. A proteasome defect in lymphoid cells
of NOD mice impairs the presentation of self antigens and
increases the susceptibility of these cells to TNF-![]() –induced apoptosis.
Here, we examine the hypothesis that induction of TNF-
–induced apoptosis.
Here, we examine the hypothesis that induction of TNF-![]() expression combined with
reeducation of newly emerging T cells with self
antigens can interrupt autoimmunity. Hyperglycemic NOD
mice were treated with CFA to induce TNF-
expression combined with
reeducation of newly emerging T cells with self
antigens can interrupt autoimmunity. Hyperglycemic NOD
mice were treated with CFA to induce TNF-![]() expression and were
exposed to functional complexes of MHC class I molecules and
antigenic peptides either by repeated injection of MHC class
I matched splenocytes or by transplantation of islets from
nonautoimmune donors. Hyperglycemia was controlled in
animals injected with splenocytes by administration of
insulin or, more effectively, by implantation of
encapsulated islets. These interventions reversed the
established ß cell–directed autoimmunity and
restored endogenous pancreatic islet function to such an extent
that normoglycemia was maintained in up to 75% of animals after
discontinuation of treatment and removal of islet transplants.
A therapy aimed at the selective elimination of
autoreactive cells and the reeducation of T cells,
when combined with control of glycemia, is thus able
to effect an apparent cure of established type 1
diabetes in the NOD mouse.
expression and were
exposed to functional complexes of MHC class I molecules and
antigenic peptides either by repeated injection of MHC class
I matched splenocytes or by transplantation of islets from
nonautoimmune donors. Hyperglycemia was controlled in
animals injected with splenocytes by administration of
insulin or, more effectively, by implantation of
encapsulated islets. These interventions reversed the
established ß cell–directed autoimmunity and
restored endogenous pancreatic islet function to such an extent
that normoglycemia was maintained in up to 75% of animals after
discontinuation of treatment and removal of islet transplants.
A therapy aimed at the selective elimination of
autoreactive cells and the reeducation of T cells,
when combined with control of glycemia, is thus able
to effect an apparent cure of established type 1
diabetes in the NOD mouse.
|
Introduction |
Autoimmune destruction of pancreatic ß cells is more than
90% complete by the time hyperglycemia becomes clinically evident
in individuals with type 1 (insulin-dependent) diabetes mellitus.
Prevention of this disease would therefore optimally require
arrest of autoimmunity in the prehyperglycemic phase. After
hyperglycemia is established, therapies based on islet cell
replacement are necessary to restore physiological control of
blood glucose. Although islet transplantation has been successful
in mice, rats, and nonhuman primates with chemically
induced diabetes, sustained survival of allogeneic
islet grafts is infrequently observed in spontaneous
diabetic hosts such as the NOD (nonobese diabetic)
mouse, BB (BioBreeding) rat, and diabetic humans (1-5).
Thus, allogeneic or xenogeneic cellular grafts subjected to
transient ablation of donor MHC class I antigen expression
(6)
— achieved either with the use of "masking"
Ab’s to MHC class I molecules or by deletion of
the ß2-microglobulin (ß2M)
gene — are capable of permanent engraftment in
nonautoimmune recipients, but are minimally protected from recurrent
ß cell autoimmunity in NOD mice (7,
8).
The immune mechanisms of islet graft rejection and
recurrent autoimmunity appear distinct, and protective
interventions targeted at these two pathways of islet
destruction are nonoverlapping in effectiveness.
Subsets of antigen-presenting cells and T cells of NOD mice
that progress to hyperglycemia exhibit a decrease in the
production of LMP2, a catalytic subunit of the
proteasome, after about 5–6 weeks of age (9,
10).
This defect is accompanied by deficient generation of
peptides from endogenous proteins for display on the
cell surface by MHC class I molecules, a process that
is important for T cell memory and tolerance to self antigens
(11,
12)
and is impaired in various human and murine autoimmune diseases
(11,
13,
14).
The proteasome also contributes to the processing and
activation of NF-![]() B (15-17),
a transcription factor that regulates the expression
of genes that contribute to cytokine generation,
lymphocyte maturation, protection from TNF-
B (15-17),
a transcription factor that regulates the expression
of genes that contribute to cytokine generation,
lymphocyte maturation, protection from TNF-![]() –induced apoptosis,
and promotion of the processing of antigens for presentation
by MHC class I molecules (18-20).
The proteasome defect in NOD mice affects lymphoid
maturation as a result, at least in part, of impaired
activation of NF-
–induced apoptosis,
and promotion of the processing of antigens for presentation
by MHC class I molecules (18-20).
The proteasome defect in NOD mice affects lymphoid
maturation as a result, at least in part, of impaired
activation of NF-![]() B.
B.
In normal humans and other mammals, the continuous expression
of MHC class I molecules by peripheral cells, including
islet cells (21),
maintains peripheral tolerance in the context of properly
selected lymphocytes (12,
22).
Interruption of exposure to complexes of self peptides
and MHC class I molecules results in the aberrant
selection of CD8+ T cells that exhibit an increased
sensitivity to apoptosis (23).
Diabetic humans and NOD mice that progress to diabetes
thus manifest a paucity of memory cells and an
increased susceptibility of T cells to apoptosis, traits
that may be secondary, in part, to improper presentation of
self antigens by MHC class I molecules and ineffective T cell
selection (10,
11).
On the basis of the view that islet transplantation into
hyperglycemic NOD mice will require both the
prevention of transplant rejection and the elimination
of autoimmune T cells, we sought to combine two
strategies to achieve these goals. To bypass graft rejection,
we used donor islets from C57BL/6 mice in which the ß2M
gene was deleted. MHC class I proteins are re-expressed on
graft cells within 24 to 72 hours after
transplantation as a result of reconstitution with
host ß2M present in plasma (6,
24,
25).
The re-expression of donor MHC class I antigens is
important because it is necessary for the development and maintenance
of peripheral tolerance.
With regard to interruption of autoimmunity, we hypothesized
that the lineage-specific defects both in peptide
presentation by MHC class I molecules and in the
processing and activation of NF-![]() B might be important in the
pathogenesis of diabetes in NOD mice. The NF-
B might be important in the
pathogenesis of diabetes in NOD mice. The NF-![]() B defect in the affected
lineages on NOD mice is accompanied by an increased
sensitivity of these cells to TNF-
B defect in the affected
lineages on NOD mice is accompanied by an increased
sensitivity of these cells to TNF-![]() –induced apoptosis in
vitro (10).
The increased susceptibility to apoptosis of
misselected T cells that result from improper education
by MHC class I peptide complexes suggested that the production
of TNF-
–induced apoptosis in
vitro (10).
The increased susceptibility to apoptosis of
misselected T cells that result from improper education
by MHC class I peptide complexes suggested that the production
of TNF-![]() in
vivo might promote the selective death of such poorly
educated lineages (10,
26,
27).
Furthermore, treatment of diabetic NOD mice or BB rats
with CFA, an inducer of TNF-
in
vivo might promote the selective death of such poorly
educated lineages (10,
26,
27).
Furthermore, treatment of diabetic NOD mice or BB rats
with CFA, an inducer of TNF-![]() production, both impairs the
transfer of disease by T cells from these animals to
naive hosts (28-31)
as well as prolongs the survival of syngeneic islet
grafts in spontaneously diabetic hosts (2,
32).
We therefore hypothesized that CFA treatment might
eliminate, at least temporarily, the autoreactive lymphoid cells
of NOD mice by promoting their apoptosis, in part through the
induction of TNF-
production, both impairs the
transfer of disease by T cells from these animals to
naive hosts (28-31)
as well as prolongs the survival of syngeneic islet
grafts in spontaneously diabetic hosts (2,
32).
We therefore hypothesized that CFA treatment might
eliminate, at least temporarily, the autoreactive lymphoid cells
of NOD mice by promoting their apoptosis, in part through the
induction of TNF-![]() .
.
Thus, we both treated hyperglycemic NOD recipients with CFA,
seeking to eliminate autoreactive lymphoid lineages, and
transplanted islets from ß2M-deficient
donors under the kidney capsule of these animals,
seeking to generate graft-specific tolerance. These
interventions resulted in the marked reduction or
apparent elimination of ongoing ß cell–directed autoimmunity.
Unexpectedly, the long-term reappearance of endogenous ß
cell function was also observed in the pancreatic islets
of the previously hyperglycemic hosts.
|
Methods |
Animals. Female NOD mice from Taconic Farms
(Germantown, New York, USA) and C57BL/6J (C57) mice
from The Jackson Laboratory (Bar Harbor, Maine, USA)
were maintained under pathogen-free conditions. NOD
mice were screened for the onset of diabetes by monitoring body
weight and blood glucose; they were diagnosed as diabetic when
two consecutive blood glucose concentrations exceeded 400 mg/dl.
Before experimental treatments, diabetic NOD mice were maintained
for 7–20 days on daily injections of 1.0–1.5 U
of NPH human insulin per 100 g of body weight, thereby preventing
immediate death and maintaining blood sugar concentration
between 200 and 700 mg/dl. The use of such severely
diabetic mice, relatively late after disease onset,
ensured that endogenous pancreatic islets were
completely obliterated before the initiation of experiments.
Splenocyte donors included normal C57 mice, C57 mice
(C57 ß2M–/–)
in which the ß2M gene was disrupted, C57
mice (C57 ß2M–/–,
TAP1–/–) in which both
the ß2M and Tap1 genes were
disrupted, and MHC class II–/–
mice (C57 class II–/–)
in which the I-A gene was disrupted and the E locus of
MHC class II was not expressed because of an
endogenous defect in the C57 strain (Taconic Farms). Splenocytes
(9 x 106) were injected into NOD recipients
through the tail vein twice a week. CFA (Difco
Laboratories, Detroit, Michigan, USA) was freshly
mixed with an equal volume of physiological saline and
injected (50 µl) into each hind-foot pad at the time
of islet transplantation or after the first splenocyte injection.
Islet transplantation. Islets were isolated
from donor C57 mice or 6- to 8-week-old prediabetic
female NOD mice. Density gradient centrifugation followed
by hand picking of islets ensured that both preparations were
highly enriched in islets and had an accurate determination of
islet number. For transplantation, 500–600 freshly isolated
islets were grafted beneath the left renal capsule of
each diabetic NOD recipient. For islet encapsulation,
900–1,100 islets were enclosed in alginate
spheres, which were then surgically inserted into the
peritoneal cavity of diabetic NOD mice. Transplantation was
considered successful if the nonfasting blood concentration of
glucose returned to normal (<200 mg/dl) within 24 hours after
surgery. The glucose concentration of orbital blood was monitored
three times a week after transplantation with a Glucometer Elite
instrument (Bayer Corp, Pittsburgh, Pennsylvania, USA). Body
weight was also monitored three times a week. Islet grafts were
considered to have been rejected if the blood glucose
concentration increased to more than 250 mg/dl on two
occasions. Recipients that rejected islet grafts were
killed for histological examination and
flow-cytometric studies. To assess the contribution of endogenous
pancreatic islets to the control of blood sugar
concentration, we removed subrenal islet transplants
by nephrectomy. Similarly, islets encapsulated in
alginate spheres, which were approximately 0.2–0.5
cm in diameter, were removed from the peritoneal cavity
by direct visualization under a dissecting microscope. Histological
analysis of the pancreata and islet grafts was performed
by staining with hematoxylin and eosin for evaluation of
lymphocytic infiltrates and with aldehyde-fuchsin for islet insulin
content. The entire pancreas from splenic to duodenal stomach
attachments was removed, fixed, embedded longitudinally in
a paraffin block, and subjected to serial sectioning (10 µm).
Flow cytometry. Spleens were removed and
gently minced on a stainless steel sieve. Cell
suspensions were rendered free of red blood cells by
exposure to a solution containing 0.83% NH4Cl. The splenocytes
were stained with mouse mAb’s (PharMingen, San Diego,
California, USA) to CD8 (FITC-labeled), to CD62L
(phycoerythrin-labeled [PE-labeled]), to CD95
(PE-labeled), or to CD45RB (PE-labeled), and were
analyzed (>10,000 cells per sample) the same day with
an Epics Elite flow cytometer. Spleen cells were incubated for
24 hours in the absence or presence of TNF-![]() (20 ng/ml), after
which apoptotic cells were detected by flow cytometry with
FITC-conjugated annexin V. Apoptotic cells were defined as
cells positive for both propidium iodide (PI) and annexin V
staining; numbers within the upper quadrants represent the corresponding
percentages of cells.
(20 ng/ml), after
which apoptotic cells were detected by flow cytometry with
FITC-conjugated annexin V. Apoptotic cells were defined as
cells positive for both propidium iodide (PI) and annexin V
staining; numbers within the upper quadrants represent the corresponding
percentages of cells.
Adoptive transfer. Adoptive transfer was
performed as described (29).
Recipient male NOD mice, 4–8 weeks of age, were
irradiated (790 rads) with a 137Cs source
and injected intravenously within 2 hours of
irradiation with donor splenocytes (2 x 107 viable
cells) suspended in 0.25 ml of serum-free medium. Diabetic
spleen cell donors were female NOD mice that typically
had exhibited blood sugar concentrations of greater
than 400 mg/dl for at least 3 weeks. Multiple diabetic
donor spleens were pooled to produce sufficient cells
for all of the hosts in a given experiment.
Statistics. Exact algorithm P values
were calculated in some instances with multiple
comparisons corrected by the number of tested variables. A
P value less than 0.05 was considered statistically
significant.
|
Results |
CFA treatment and islet transplantation in NOD mice.
Hosts for the transplantation experiments were severely diabetic
female NOD mice, usually more than 20 weeks of age, that
had exhibited blood glucose concentrations of greater
than 400 mg/dl for at least 7 days and had been
treated by daily administration of insulin to prevent
death. Islet transplants were placed unilaterally under
the kidney capsule to facilitate nonlethal removal and histological
examination. Islets from 6- to 8-week-old prediabetic NOD
females (recipient group A) or from normal C57 mice (recipient
group B) were rapidly rejected by diabetic NOD recipients
(Table 1,
Figure 1a).
Although C57 donor islets with transient ablation of
MHC class I expression survive indefinitely in nonautoimmune
diabetic hosts (6),
the survival time of islets from ß2M–/–
C57 mice in diabetic NOD females (group C) was only about
three times that of normal C57 islets. As expected
from previous observations (2,
32),
treatment with CFA prolonged the survival of syngeneic islet
grafts in diabetic NOD hosts (group D) but had a minimal effect
on the survival of C57 islets (group E), which were uniformly
rejected by 11 days after transplantation. However, the
combination of ß2M–/–
C57 islet transplants with CFA treatment resulted in
sustained (>129 days) normoglycemia in 5 of 14
diabetic NOD hosts (group F). Although the duration of
hyperglycemia before initiation of therapy varied between 7
and 20 days, the length of this interval was not significantly
related to the duration of sustained normoglycemia after
treatment (data not shown). The animals that exhibited
sustained normoglycemia also demonstrated progressive
weight gain, similar to that apparent in NOD female
cohorts who never became diabetic (data not shown). With
normalization of blood sugar concentration as a measure of
treatment success, the success of ß2M–/–
C57 islet transplantation together with CFA administration
was significantly different from that apparent with
the other groups combined but did not differ from that
of NOD islet transplantation together with CFA
treatment.
|
|
After the recurrence of hyperglycemia in the NOD mice that had
been treated with CFA and syngeneic (NOD) islet
transplants, the kidney containing the islet graft was
examined histologically. Marked lymphocytic
infiltration was apparent under the kidney capsule at
the site of transplantation, a characteristic of recurrent
autoimmune disease (Figure 1b,
group D); moreover, no intact islets were detected in
the pancreas, although islet remnants, largely
obscured by dense pockets of infiltrating lymphocytes,
were evident. Similar histological characteristics of
both the transplant site and pancreas were apparent in diabetic
NOD mice that had received CFA and islet grafts from C57
donors (Figure 1b,
group E). Unexpectedly, for all five NOD mice with long-term
normoglycemia after receiving ß2M–/–
C57 islets and CFA treatment, no surviving islet grafts
were detected under the kidney capsule when the
animals were killed at more than 129 days after
transplantation (Figure 1b,
group F). In contrast, the pancreas of each of these
five recipients exhibited well-formed islets that
appeared completely granulated when stained by
aldehyde-fuchsin. The islets were free of lymphocytes or
lymphocytes were present only circumferentially; this latter
pattern of lymphocyte accumulation, with lymphocytes
surrounding but not invading the islets, has been
associated with nonprogressive or interrupted ß cell
autoimmunity (33).
The return to normoglycemia in the absence of
detectable transplanted islet tissue, together with
the presence of islets in a pancreas largely devoid of
infiltrating lymphocytes, suggested not only that autoimmunity
had been interrupted but that the function of endogenous ß
cells had been restored.
Restoration of near-normal pancreatic islet histology was
observed only in the diabetic NOD mice that received
both ß2M–/–
islet grafts and CFA treatment (Figure 2).
Pancreatic islets were thus not detected in any
diabetic NOD mice treated with CFA and syngeneic NOD
islets; the persistence of normoglycemia in such
recipients appeared solely due to the transplanted islets, which
always exhibited invasive insulitis (Figure 1b,
Figure 2).
Thus, treatment with CFA together with syngeneic NOD islets may
have slowed disease recurrence, but persistent autoimmunity remained.
|
The relative contributions of restored endogenous pancreatic
islets and transplanted islets to the maintenance of
normoglycemia in NOD mice treated with CFA and islet
grafts from ß2M–/–
C57 donors were assessed by removal of the kidney
containing the islet transplant after 120 days of
normoglycemia in a second group of five animals. All
five mice remained normoglycemic after nephrectomy
until they were killed 3–60 days later (Figure 3).
Histological analysis of the kidneys that received the
grafts revealed a complete loss of identifiable islet structures.
In contrast, the pancreata of all five recipients contained
well-formed islets either without lymphocytic infiltration
or with circumferentially distributed lymphocytes
only. Normoglycemia after nephrectomy was thus
maintained solely by endogenous pancreatic islets. In
contrast, nephrectomies performed during the posttransplantation
period of normoglycemia (day 62, day 85) in two mice who
had received CFA plus syngeneic NOD islets resulted in
a rapid return to hyperglycemia (data not shown),
demonstrating that the control of blood sugar in this
treatment group was mediated solely by the
transplanted islet tissue.
|
We also transplanted diabetic NOD females with islets from C57
mice in which the genes for both ß2M and TAP1
had been deleted. Together with TAP2, TAP1 mediates
the transport of endogenous peptides from the cytosol
into the lumen of the endoplasmic reticulum for their
assembly with MHC class I molecules (34).
Islet cells from these mice are more permanently defective in
presentation of self antigens by MHC class I than are those from
ß2M–/–
mice. Transplantation of ß2M–/–,
TAP1–/– C57 islets
combined with injection of CFA resulted in the return
of hyperglycemia within 14 days in five of six animals
(group G); histological examination of the pancreata revealed
a pattern typical of that for untreated diabetic NOD mice
(Table 1,
Figure 2).
Thus, a transient interruption of peptide presentation
by donor MHC class I molecules is essential for the
abrogation of autoimmunity, whereas a sustained interruption
of this process prevents the reestablishment of tolerance
and the restoration of endogenous pancreatic islet
integrity.
CFA treatment and splenocyte injection in NOD mice.
Given that the restoration of normoglycemia in the diabetic NOD
mice treated with CFA and ß2M–/–
C57 islets did not depend on the continuing secretion
of insulin by the islet grafts, we next investigated
whether C57 donor cell types other than islets might
serve a similar therapeutic role. Nine diabetic NOD
mice were treated with a single bilateral injection of
CFA followed by a 40-day regimen of biweekly intravenous injections
of C57 splenocytes. These lymphoid cells express both
MHC class I and class II proteins and survive only transiently
in NOD hosts because of graft rejection (data not shown).
Repeat injections of splenocytes ensure that the host
is continuously exposed to intact antigen presentation
complexes on the surface of these cells. The
recipients were monitored for hyperglycemia every 3 or
4 days, and insulin was administered daily unless normoglycemia
returned. A control group of four diabetic NOD mice
received daily insulin injections only. All four control mice
died on or before day 25 of the experimental period as a
result of poor control of blood glucose and consequent ketosis
and cachexia (Figure 4a).
In contrast, seven of the nine mice injected with CFA
and C57 splenocytes were alive after 40 days, and
three of these animals had become normoglycemic and insulin independent
(Figure 4b).
The pancreata of control (insulin treatment only) mice
exhibited pronounced lymphocytic infiltrates that obscured
any remaining islet structures (Figure 4c).
The pancreata of the four NOD mice treated with CFA
and C57 splenocytes that remained alive but
hyperglycemic and insulin dependent revealed a marked
decrease (relative to control animals) in the number of
lymphoid infiltrates, which were located either circumferentially
or adjacent to the infrequent islet structures (Figure 4d).
On killing of each of the three NOD mice treated with CFA
and C57 splenocytes that maintained normoglycemia
after discontinuation of insulin injections, the
pancreata exhibited abundant islets that were free of
invasive lymphocytes or were associated only with
circumferential lymphocytes (Figure 4e).
Thus, treatment with CFA combined with repeated
exposure to C57 lymphocytes resulted in complete
reversal of diabetes in approximately 30% of NOD
recipients and partially restored ß cell function in
an additional approximately 40% of recipients.
|
Influence of glycemic control on restoration of
endogenous islet function. The reversal of diabetes in
NOD mice by CFA and repeated exposure to C57
splenocytes indicated that restoration of endogenous islet
function is achievable without islet transplantation and despite
the poor glycemic control attained by insulin injections. The
beneficial influence of glycemic control on the growth, survival,
and function of cultured islets, as well as of transplanted islets
in nonautoimmune settings, has been demonstrated (35,
36).
To determine whether the restoration of endogenous ß cell
function could be achieved more consistently with better control
of blood glucose, we replaced insulin injections with the
intraperitoneal implantation of alginate-encapsulated C57 mouse
islets. Alginate encapsulation prevents direct contact between
donor endocrine cells and host T cells, and such grafts have
been shown to provide near-normal glycemic control for 40
to 50 days in approximately 70–80% of autoimmune NOD recipients
(37).
Almost all diabetic NOD mice that received
alginate-encapsulated C57 islets exhibited improved
glucose regulation or normoglycemia. The alginate
spheres were removed 40–50 days after implantation, and
blood glucose concentration was monitored (Table 2).
The seven mice treated only with alginate-encapsulated
islets (group A), the six mice that received a single
bilateral injection of CFA (group B), and the three
mice treated with biweekly injections of C57
splenocytes (data not shown) all exhibited a rapid return to
hyperglycemia and early death after removal of the implants (Table
2,
Figure 5).
The pancreata of NOD mice that received only
alginate-encapsulated islets revealed no sign of intact islets
or of lymphoid infiltrates (data not shown). The pancreata of
NOD hosts treated with CFA and alginate-encapsulated islets exhibited
marked invasive insulitis that obscured islet structures (Figure
6).
In contrast, seven of the nine (78%) diabetic NOD mice
that received CFA and C57 splenocytes (group C) remained normoglycemic
for more than 40 days (until killing) after removal of
the alginate-encapsulated islets (Figure 5,
Table 2).
The pancreata of these animals contained large islets
with circumferentially distributed lymphocytes (Figure
6).
The islet mass after at least 80 days of disease
reversal was estimated at approximately 50% of the
original value. The pancreata from control BALB/c mice
contained approximately 25–35 islets; the pancreata from
successfully treated NOD mice contained approximately 12–20
islets with serial histological sections. Thus, maintenance
of normoglycemia during the treatment period increased the
percentage of diabetic mice cured of hyperglycemia.
|
|
|
Role of TNF-![]() in treatment
outcome. We attempted to identify features of the
successful treatment regimens that are critical to a
positive outcome. We had used CFA to induce the
endogenous production of TNF-
in treatment
outcome. We attempted to identify features of the
successful treatment regimens that are critical to a
positive outcome. We had used CFA to induce the
endogenous production of TNF-![]() (31).
The importance of TNF-
(31).
The importance of TNF-![]() in treatment success was
therefore investigated by the intravenous
administration of a rat IgG1 mAb to this cytokine (clone
MP6-X73; Accurate Chemical & Scientific Corp., Westbury,
New York, USA) at a dose of 1.5 mg/day for the first 10
days in diabetic NOD hosts treated with C57
splenocytes, CFA, and alginate-encapsulated islets.
All five NOD mice so treated exhibited a rapid return
to hyperglycemia on removal of the alginate-encapsulated islets
50–70 days after transplantation (Table 2,
group F), consistent with the notion that TNF-
in treatment success was
therefore investigated by the intravenous
administration of a rat IgG1 mAb to this cytokine (clone
MP6-X73; Accurate Chemical & Scientific Corp., Westbury,
New York, USA) at a dose of 1.5 mg/day for the first 10
days in diabetic NOD hosts treated with C57
splenocytes, CFA, and alginate-encapsulated islets.
All five NOD mice so treated exhibited a rapid return
to hyperglycemia on removal of the alginate-encapsulated islets
50–70 days after transplantation (Table 2,
group F), consistent with the notion that TNF-![]() plays an obligatory role
in the beneficial effect of CFA. This effect of the mAb to
TNF-
plays an obligatory role
in the beneficial effect of CFA. This effect of the mAb to
TNF-![]() was
specific, given that administration of a rat IgG1 mAb
to the human T cell receptor Vß1 chain (clone BL37.2;
American Type Culture Collection, Rockville, Maryland, USA)
at a dose of 1.5 mg/day for 10 days did not affect the success
of treatment with C57 splenocytes and CFA (data not shown).
Direct administration of TNF-
was
specific, given that administration of a rat IgG1 mAb
to the human T cell receptor Vß1 chain (clone BL37.2;
American Type Culture Collection, Rockville, Maryland, USA)
at a dose of 1.5 mg/day for 10 days did not affect the success
of treatment with C57 splenocytes and CFA (data not shown).
Direct administration of TNF-![]() to diabetic hosts was not
feasible because of the prohibitive cost.
to diabetic hosts was not
feasible because of the prohibitive cost.
We next investigated whether the production of TNF-![]() in diabetic NOD
mice treated with CFA results in the selective elimination of
autoreactive lymphoid cells first by examining the susceptibility
of lymphocytes from successfully treated animals to TNF-
in diabetic NOD
mice treated with CFA results in the selective elimination of
autoreactive lymphoid cells first by examining the susceptibility
of lymphocytes from successfully treated animals to TNF-![]() –induced cell
death in vitro. As shown previously (26,
27),
incubation of normal C57 spleen cells with TNF-
–induced cell
death in vitro. As shown previously (26,
27),
incubation of normal C57 spleen cells with TNF-![]() in vitro had no effect on
cell viability; for the animal shown in Figure 7a,
the proportion of apoptotic cells was 0.01% for
splenocytes incubated in the absence or presence of TNF-
in vitro had no effect on
cell viability; for the animal shown in Figure 7a,
the proportion of apoptotic cells was 0.01% for
splenocytes incubated in the absence or presence of TNF-![]() . In contrast, exposure of
splenocytes from an untreated 20-week-old NOD female
to TNF-
. In contrast, exposure of
splenocytes from an untreated 20-week-old NOD female
to TNF-![]() in
vitro increased the proportion of apoptotic cells from
0.03 to 38.3%. Such an increased sensitivity to TNF-
in
vitro increased the proportion of apoptotic cells from
0.03 to 38.3%. Such an increased sensitivity to TNF-![]() –induced apoptosis in
vitro was no longer evident with spleen cells derived
from NOD mice cured of diabetes; thus, splenocytes
from a NOD female successfully treated with both CFA
and C57 splenocytes (Table 2,
group C) exhibited 23.2 and 23.9% apoptosis in the
absence and presence of TNF-
–induced apoptosis in
vitro was no longer evident with spleen cells derived
from NOD mice cured of diabetes; thus, splenocytes
from a NOD female successfully treated with both CFA
and C57 splenocytes (Table 2,
group C) exhibited 23.2 and 23.9% apoptosis in the
absence and presence of TNF-![]() , respectively (Figure 7a).
Successful therapy generated a subpopulation of
nonpathologic but TNF-
, respectively (Figure 7a).
Successful therapy generated a subpopulation of
nonpathologic but TNF-![]() –resistant T cells
that exhibited an increased tendency to undergo cell death in
culture (Figure 7a).
Disease reversal, even 210 days after cessation of
treatment, was thus associated with the persistent elimination
of TNF-
–resistant T cells
that exhibited an increased tendency to undergo cell death in
culture (Figure 7a).
Disease reversal, even 210 days after cessation of
treatment, was thus associated with the persistent elimination
of TNF-![]() –sensitive
T cells, a population of cells with a demonstrated
ability to play a role in disease (29,
30).
The permanent elimination of these formerly abundant TNF-
–sensitive
T cells, a population of cells with a demonstrated
ability to play a role in disease (29,
30).
The permanent elimination of these formerly abundant TNF-![]() –sensitive lymphoid
cells, presumably in response to TNF-
–sensitive lymphoid
cells, presumably in response to TNF-![]() (and, perhaps, to other
CFA-induced cytokines), was observed uniformly in
successfully treated diabetic NOD mice. Similar
complete and stable elimination of TNF-
(and, perhaps, to other
CFA-induced cytokines), was observed uniformly in
successfully treated diabetic NOD mice. Similar
complete and stable elimination of TNF-![]() –sensitive cells
at various times after treatment has been observed in more
than 20 NOD mice.
–sensitive cells
at various times after treatment has been observed in more
than 20 NOD mice.
|
We also examined the effect of TNF-![]() on the pathogenesis of
autoimmune diabetes in adoptive transfer experiments.
Young recipient NOD males were subjected to
irradiation followed by an intravenous injection of
donor splenocytes either from newly diabetic NOD females
or from NOD mice with long-term normoglycemia due to treatment
with CFA and C57 splenocytes. The onset of diabetes was
observed in all recipients by day 15 after the transfer of
diabetic mouse cells that were injected either immediately after
isolation or after control culture for 24 hours (Figure 7b,
left panel). In contrast, four of the five recipients of diabetic
mouse cells that had been cultured with TNF-
on the pathogenesis of
autoimmune diabetes in adoptive transfer experiments.
Young recipient NOD males were subjected to
irradiation followed by an intravenous injection of
donor splenocytes either from newly diabetic NOD females
or from NOD mice with long-term normoglycemia due to treatment
with CFA and C57 splenocytes. The onset of diabetes was
observed in all recipients by day 15 after the transfer of
diabetic mouse cells that were injected either immediately after
isolation or after control culture for 24 hours (Figure 7b,
left panel). In contrast, four of the five recipients of diabetic
mouse cells that had been cultured with TNF-![]() for 24 hours
before adoptive transfer remained normoglycemic for at least
40 days after cell injection (Figure 7b,
middle panel). Furthermore, the four NOD hosts each
injected with splenocytes from a different NOD female
that had experienced reversal of autoimmunity for more
than 120 days failed to develop disease during
observation periods of more than 60 days (Figure 7b,
right panel). TNF-
for 24 hours
before adoptive transfer remained normoglycemic for at least
40 days after cell injection (Figure 7b,
middle panel). Furthermore, the four NOD hosts each
injected with splenocytes from a different NOD female
that had experienced reversal of autoimmunity for more
than 120 days failed to develop disease during
observation periods of more than 60 days (Figure 7b,
right panel). TNF-![]() –resistant NOD
splenocytes, enriched either in vitro by direct
exposure of cells to TNF-
–resistant NOD
splenocytes, enriched either in vitro by direct
exposure of cells to TNF-![]() or in vivo by
disease reversal, are thus incapable of transferring disease
to naive hosts.
or in vivo by
disease reversal, are thus incapable of transferring disease
to naive hosts.
Role of MHC class I peptide complexes in T cell
selection and treatment outcome. Disease reversal in
diabetic NOD mice required treatment with both CFA and
cells that express MHC class I peptide complexes. Only
two of six (33%) diabetic NOD mice that received CFA and biweekly
injections of splenocytes from ß2M–/–,
TAP1–/– C57 donors
remained normoglycemic after removal of
alginate-encapsulated islets (Table 2,
group D). The pancreata of the four animals that
became hyperglycemic after removal of the alginate
spheres contained no granulated islets and only a few
visible islet structures, which were invaded and
obscured by lymphocytic infiltrates (Figure 6).
In contrast, 8 of 11 (73%) diabetic NOD mice treated
with CFA and splenocytes from C57 donors lacking MHC
class II protein expression remained normoglycemic
after removal of the alginate-encapsulated islets (Table
2,
group E); the pancreata of these animals contained large
islets that exhibited only moderate lymphocytic accumulation
at the periphery (Figure 6).
Interruption of antigen presentation by MHC class I skews the
T cell repertoire to an overabundance of naive cells, a
consistent trait of diabetes-prone NOD mice and humans
(38-40).
Improper T cell selection secondary to interruption of
antigen presentation by MHC class I results in
overexpression of CD95 by CD8+ T cells as
well as an increase in the abundance of cells with naive cell
markers such as CD62L+ and CD45RBhigh (12,
23).
To investigate whether therapeutic reversal of NOD
mouse diabetes was associated with a change in naive T
cell selection, we subjected splenocytes to
flow-cytometric analysis. Flow cytometry was performed 5 to
26 days after removal of the alginate-encapsulated islets and
termination of therapy.
Untreated NOD mice exhibited the expected increases in the
abundance of naive CD8+CD45RBhigh,
CD8+CD62L+, and CD8+CD95+
cells compared with C57 animals (Figure 7c).
In contrast, in NOD female mice that were successfully
treated with alginate-encapsulated islets, CFA, and
administration of C57 splenocytes, the percentages of
each of these cell populations were reduced to normal or near-normal
values. The abnormally high numbers of CD8+CD45RBhigh,
CD8+CD62L+, and CD8+CD95+
cells remained increased in diabetic NOD females
treated with alginate-encapsulated islets and CFA, either
alone or together with administration of ß2M–/–,
TAP1–/– C57 splenocytes.
Data are means plus or minus SEM of values from at
least five mice per group. Exact algorithm P
values were calculated for comparisons of each cell population
between groups C, E, and F versus groups A, B, and D: P
= 0.001 for CD8+CD45RBhigh cells, P
= 0.01 for CD8+CD62L+ cells, and P =
0.05 for CD8+CD95+ cells. Despite the fact
that many comparisons were performed, the P
value remained less than 0.05 even when multiplied by
the three comparisons. These data showed that the T
cell reselection apparent in successfully treated NOD
mice was secondary to reexposure to complexes of MHC
class I molecules and self peptides. The normalization of T
cell phenotype did not require reexposure to MHC class
II–peptide complexes, given that the
administration together with CFA and alginate-encapsulated
islets of MHC class II–/– splenocytes
was as effective as was that of normal C57 splenocytes.
|
Discussion |
We have demonstrated the effectiveness of a novel therapy for
the correction of established autoimmune diabetes in the
NOD mouse. Three aspects of this treatment regimen
appear to operate in parallel and in a synergistic
manner: (a) Injection of CFA, and the consequent
induction of TNF-![]() , results in the elimination
of TNF-
, results in the elimination
of TNF-![]() –sensitive cells, which
have been shown previously to transfer existing
disease (28-31);
(b) the introduction of functional MHC class I peptide
complexes expressed on the surface of either normal
islet cells or normal lymphocytes results in partial
but stable reselection of the T cell population of the NOD
host, leading to an increase in the abundance of long-term memory
T cells (6,
12,
23);
and (c) suppression of hyperglycemia, although not
obligatory, promotes the functional restoration of
endogenous ß cells or their precursors.
–sensitive cells, which
have been shown previously to transfer existing
disease (28-31);
(b) the introduction of functional MHC class I peptide
complexes expressed on the surface of either normal
islet cells or normal lymphocytes results in partial
but stable reselection of the T cell population of the NOD
host, leading to an increase in the abundance of long-term memory
T cells (6,
12,
23);
and (c) suppression of hyperglycemia, although not
obligatory, promotes the functional restoration of
endogenous ß cells or their precursors.
We propose that continuous or repeated exposure to parenchymal
or lymphoid cells expressing MHC class I molecules and self
peptides initiates the reeducation of host T cells, which
was apparent in CFA-treated hosts from the loss of
cells with an increased sensitivity to TNF-![]() –induced apoptosis and
from the restoration of a cell surface phenotype
characteristic of long-term memory cells. This
reeducation resulted in the establishment of long-term
tolerance, as demonstrated by the elimination of both
recurrent hyperglycemia and invasive insulitis. Treatment of
NOD mice with severe hyperglycemia and islet destruction resulted
in the reappearance of pancreatic insulin-secreting cells
and normoglycemia. The rate of pancreatic ß cell
proliferation is increased during the active phase of disease
in NOD mice, and NOD islet stem cells proliferate in
culture (41).
The interruption of ß cell autoimmunity may promote
both the rescue of surviving ß cells in islets as
well as the production of new ß cells that are now
able to survive in the altered immunological milieu. The expression
of MHC class I molecules and self peptides by NOD pancreatic
ß cells (21)
may be responsible for maintenance of peripheral
tolerance after termination of disease by transient therapy.
–induced apoptosis and
from the restoration of a cell surface phenotype
characteristic of long-term memory cells. This
reeducation resulted in the establishment of long-term
tolerance, as demonstrated by the elimination of both
recurrent hyperglycemia and invasive insulitis. Treatment of
NOD mice with severe hyperglycemia and islet destruction resulted
in the reappearance of pancreatic insulin-secreting cells
and normoglycemia. The rate of pancreatic ß cell
proliferation is increased during the active phase of disease
in NOD mice, and NOD islet stem cells proliferate in
culture (41).
The interruption of ß cell autoimmunity may promote
both the rescue of surviving ß cells in islets as
well as the production of new ß cells that are now
able to survive in the altered immunological milieu. The expression
of MHC class I molecules and self peptides by NOD pancreatic
ß cells (21)
may be responsible for maintenance of peripheral
tolerance after termination of disease by transient therapy.
The application of this therapy to humans with type 1 diabetes
may be feasible. As in NOD mice, lymphocytes from type 1
diabetic humans show an increased sensitivity to TNF-![]() –induced apoptosis (10)
as well as age-related defects in MHC class I presentation of
self peptides for proper T cell selection (11,
13).
Moreover, diabetic humans continue to produce
auto-Ab’s to islet targets for several years
after the onset of frank hyperglycemia, indicating the
persistence of islet cell antigen expression. Thus, a
proportion of individuals with type 1 diabetes may possess a
ß cell mass or islet regenerative potential similar to
that of hyperglycemic NOD mice. Even if the regenerative capacity
of ß cells is exhausted, a similar immunomodulation approach
may provide a less hostile milieu for islet replacement.
–induced apoptosis (10)
as well as age-related defects in MHC class I presentation of
self peptides for proper T cell selection (11,
13).
Moreover, diabetic humans continue to produce
auto-Ab’s to islet targets for several years
after the onset of frank hyperglycemia, indicating the
persistence of islet cell antigen expression. Thus, a
proportion of individuals with type 1 diabetes may possess a
ß cell mass or islet regenerative potential similar to
that of hyperglycemic NOD mice. Even if the regenerative capacity
of ß cells is exhausted, a similar immunomodulation approach
may provide a less hostile milieu for islet replacement.
|
Acknowledgments |
This work was supported by The Iacocca Foundation. We thank Biohybrid
Technologies (J. Hayes, D. Wolf, and C. McGrath) for assistance
with islet preparation and encapsulation; S. Thompson (Bayer
Corp.) for providing surplus blood glucose monitoring strips;
M. Contant for preparation of specimens for histological analysis;
NICHD for funding to study autoimmune PDF patients; and
J. Avruch and D. Nathan for critical review of the manuscript.
|
Footnotes |
Shinichiro Ryu and Shohta Kodama contributed equally to this
work.
|
References |
This article has been cited by other articles:
|
|
S. Kodama, W. Kuhtreiber, S. Fujimura, E. A. Dale, and D. L. Faustman Islet Regeneration During the Reversal of Autoimmune Diabetes in NOD Mice Science, November 14, 2003; 302(5648): 1223 - 1227. [Abstract] [Full Text] [PDF] |
|||
|
|
G. Demirci, T. B. Strom, and X. C. Li Islet Allograft Rejection in Nonobese Diabetic Mice Involves the Common {gamma}-Chain and CD28/CD154-Dependent and -Independent Mechanisms J. Immunol., October 1, 2003; 171(7): 3878 - 3885. [Abstract] [Full Text] [PDF] |
|||
|
|
T. D. Zorina, V. M. Subbotin, S. Bertera, A. M. Alexander, C. Haluszczak, B. Gambrell, R. Bottino, A. J. Styche, and M. Trucco Recovery of the Endogenous {beta} Cell Function in the NOD Model of Autoimmune Diabetes Stem Cells, July 1, 2003; 21(4): 377 - 388. [Abstract] [Full Text] [PDF] |
|||
|
|
Y. C. Zhang, A. Pileggi, A. Agarwal, R. D. Molano, M. Powers, T. Brusko, C. Wasserfall, K. Goudy, E. Zahr, R. Poggioli, M. Scott-Jorgensen, M. Campbell-Thompson, J. M. Crawford, H. Nick, T. Flotte, T. M. Ellis, C. Ricordi, L. Inverardi, and M. A. Atkinson Adeno-Associated Virus-Mediated IL-10 Gene Therapy Inhibits Diabetes Recurrence in Syngeneic Islet Cell Transplantation of NOD Mice Diabetes, March 1, 2003; 52(3): 708 - 716. [Abstract] [Full Text] [PDF] |
|||
|
|
D. L. FAUSTMAN Reversal of Established Autoimmune Diabetes by in Situ {beta}-Cell Regeneration Ann. N.Y. Acad. Sci., June 1, 2002; 961(1): 40 - 40. [Full Text] [PDF] |
|||
|
|
A. Rabinovitch, W. L. Suarez-Pinzon, A.M. J. Shapiro, R. V. Rajotte, and R. Power Combination Therapy With Sirolimus and Interleukin-2 Prevents Spontaneous and Recurrent Autoimmune Diabetes in NOD Mice Diabetes, March 1, 2002; 51(3): 638 - 645. [Abstract] [Full Text] [PDF] |
|||
|
|
J. P. Palmer Immunomodulatory therapy of human type 1 diabetes: lessons from the mouse J. Clin. Invest., July 1, 2001; 108(1): 31 - 33. [Full Text] [PDF] |
|||
|
|
Copyright
© 2001 by the American Society for Clinical Investigation.
click
here for more information & resources
Information
about Clinical Trials
News Archives | Hotline Newsletter | News & Information | MGH Home Page